



CHAPITRE
IV
RÉNOVER LA RÉGULATION DES DÉPENSES DE PRODUITS
DE SANTÉ
Article 29
(supprimé)
Assurer une juste contribution des différents
acteurs à la régulation des produits de santé
L'article 29 a été supprimé à l'Assemblée nationale par l'amendement n° 481 de Stéphanie Rist, et déplacé en troisième partie à l'article 9 bis . En conséquence, les dispositions qu'il portait sont commentées dans le présent rapport au même article.
Article 30
Garantir
l'accès aux médicaments et l'efficience de leur prise en
charge
Cet article propose diverses mesures visant à favoriser l'accès aux médicaments, leur financement et la régulation des dépenses associées en :
- complétant les dispositifs d'accès précoce et compassionnel au médicament mis en place en 2021 ;
- réformant les modalités de financement des thérapies innovantes ;
- créant une procédure de référencement périodique et favorisant le recours aux remises pour maîtriser le prix des médicaments ;
- incitant les industriels à préserver l'accès aux médicaments d'intérêt thérapeutique majeur.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Compléter les dispositifs d'accès dérogatoire au médicament mis en place en 2021
Les dispositifs d'accès dérogatoire au médicament ont fait l'objet d'une refonte par la loi de financement de la sécurité sociale (LFSS) pour 2021 432 ( * ) . Les procédures d'accès précoce et d'accès compassionnel ont remplacé, à compter du 1 er juillet 2021, les anciennes autorisations temporaires d'utilisation (ATU) et recommandations temporaires d'utilisation (RTU). L'article 30 du PLFSS pour 2023 vise à compléter ces deux dispositifs, déjà renforcés par le PLFSS pour 2022 433 ( * ) .
1. Les dispositifs d'accès dérogatoire ont été simplifiés et renforcés depuis 2021
a) L'autorisation d'accès précoce
Remplaçant notamment l'ancienne autorisation temporaire d'utilisation dite « de cohorte » (ATUc), le dispositif d'accès précoce vise à permettre la prise en charge anticipée de médicaments destinés à traiter des maladies graves, rares ou invalidantes 434 ( * ) . Les médicaments concernés ont vocation à rejoindre une prise en charge classique par la suite. Quatre conditions cumulatives doivent être réunies :
- il n'existe pas de traitement alternatif approprié ;
- la mise en oeuvre du traitement ne peut être différée ;
- l'efficacité et la sécurité du médicament sont fortement présumées au regard des résultats d'essais thérapeutiques ;
- ces médicaments sont présumés innovants, notamment au regard d'un éventuel comparateur cliniquement pertinent.
La procédure d'accès précoce s'applique aux médicaments qui disposent d'une autorisation de mise sur le marché (AMM) mais ne sont pas encore inscrits sur la liste des spécialités remboursables, la phase de négociation avec le Comité économique des produits de santé (CEPS) pouvant s'étaler sur plusieurs mois, ou à ceux qui ne disposent pas encore d'une AMM pour l'indication considérée . Dans ce deuxième cas, l'entreprise doit s'engager à déposer une demande d'AMM dans un délai déterminé par la Haute Autorité de santé (HAS) et ne pouvant excéder deux ans 435 ( * ) .
La demande d'accès précoce est adressée par l'industriel à la HAS 436 ( * ) , qui communique sa décision dans un délai de trois mois 437 ( * ) . Pour les demandes relatives aux médicaments ne disposant pas encore d'une AMM, la décision de la HAS est prise après avis conforme de l'Agence nationale de sécurité du médicament (ANSM). L'autorisation est subordonnée au respect, par l'entreprise, d'un protocole d'utilisation thérapeutique et de recueil des données défini par la HAS et, pour les seuls médicaments ne disposant pas encore d'une AMM, l'ANSM.
Une autorisation d'accès précoce ne peut excéder une durée d'un an, toutefois renouvelable 438 ( * ) . Lorsqu'elle est accordée, le médicament est pris en charge à 100 % par l'assurance maladie dès l'autorisation 439 ( * ) . L'industriel est, en revanche, tenu de verser des remises annuelles et, après l'obtention d'une AMM, de reverser sous forme de remises supplémentaires la différence entre le chiffre d'affaires (CA) réalisé dans le cadre de l'accès précoce et celui qui aurait résulté des conditions tarifaires définies avec le CEPS après obtention de l'AMM 440 ( * ) .
En mai 2022, la HAS et l'ANSM ont tiré un premier bilan « positif » du dispositif d'accès précoce, soulignant que sur les dix premiers mois d'application, cinquante décisions ont été prises, en moyenne dans un délai de soixante jours à compter de la demande de l'industriel 441 ( * ) . Quarante d'entre elles ont conduit à l'octroi d'une autorisation, dont vingt dans les secteurs de l'oncologie et de l'hématologie.
Répartition des décisions d'autorisation d'accès précoce par aire thérapeutique, après dix mois d'application
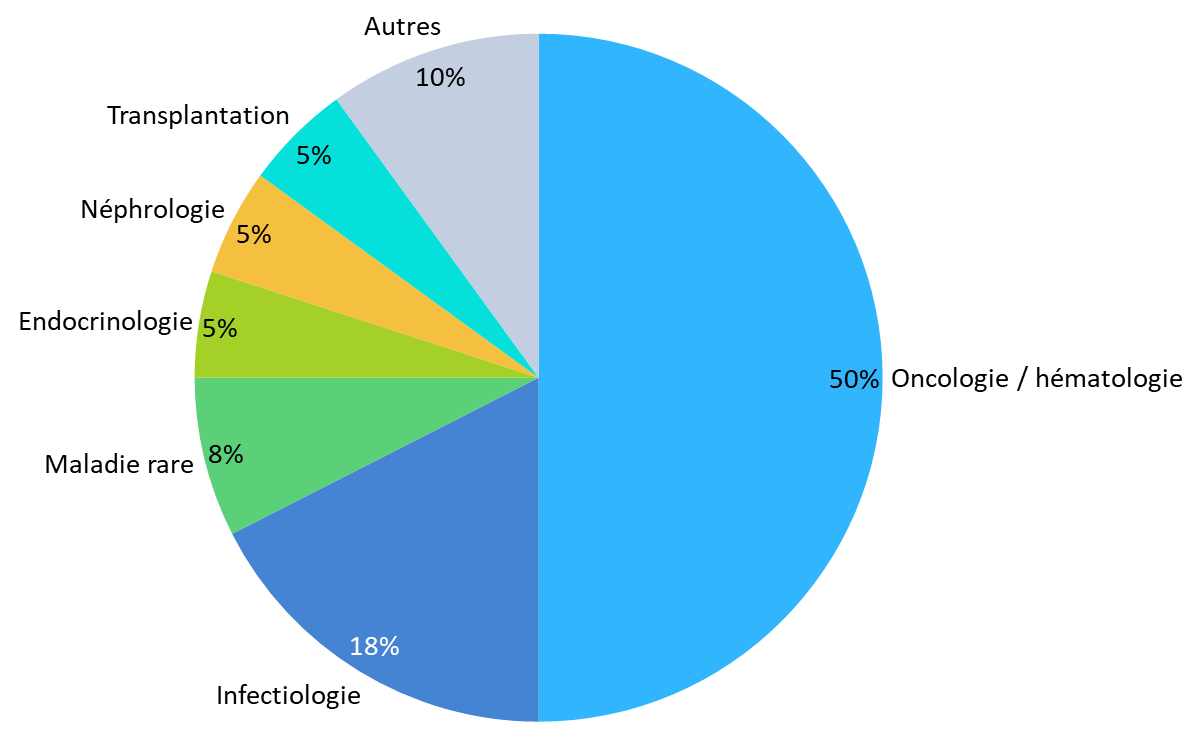
Source : Commission des affaires sociales du Sénat, d'après des données HAS et ANSM
b) L'accès compassionnel
L'accès compassionnel permet d'autoriser l'utilisation, à titre exceptionnel, d'un médicament ne faisant pas l'objet d'une recherche impliquant la personne humaine, lorsqu'aucun traitement approprié n'existe dans l'indication considérée et lorsqu'il est possible de présumer de l'efficacité et de la sécurité du médicament au regard des données cliniques disponibles 442 ( * ) . Contrairement à l'accès précoce, les médicaments visés ne sont pas nécessairement innovants : il peut s'agir de médicaments installés disposant déjà d'une AMM dans d'autres indications.
L'autorisation peut être accordée selon deux procédures :
- elle peut être sollicitée par un médecin prescripteur , au bénéfice d'un patient atteint d'une maladie grave, rare ou invalidante : l'ANSM peut alors autoriser, pour une durée maximale d'un an renouvelable, l'utilisation d'un médicament ne disposant d'aucune AMM ou d'un médicament ayant fait l'objet d'un arrêt de commercialisation et dont l'AMM ne porte pas sur l'indication sollicitée ;
- de sa propre initiative ou à la demande du ministre chargé de la santé ou de la sécurité sociale , l'ANSM peut établir pour une durée de trois ans renouvelable, un cadre de prescription relatif à un médicament faisant l'objet, pour d'autres indications, d'une AMM, afin de sécuriser une prescription non conforme à cette autorisation.
Le niveau de prise en charge est soit aligné sur celui fixé pour le CEPS dans d'autres indications, soit fixé sur la base du prix facturé aux établissements de santé ou sur une base forfaitaire annuelle par patient définie par arrêté. Comme pour l'accès précoce, l'entreprise est soumise au versement de remises 443 ( * ) .
2. L'article 30 vise à améliorer, à la marge, les dispositifs dérogatoires et leurs modalités de prise en charge
a) Des modifications mineures des modalités de liquidation des remises relatives aux médicaments en accès compassionnel
L'article 30 du PLFSS déposé améliore à la marge les modalités de prise en charge des médicaments bénéficiant de dispositifs d'accès dérogatoire.
D'abord, le 1° du I de l'article insère à l'article L. 162-16-5-2 du code de la sécurité sociale, relatif aux modalités de prise en charge des médicaments bénéficiant d'une autorisation d'accès compassionnel, un nouvel alinéa prévoyant que l'entreprise informe le CEPS, le 15 février de l'année n+1 , du CA correspondant à ce médicament ainsi que du nombre d'unités vendues, pour chacune des indications, au titre de l'année n . Ces modalités déclaratives sont identiques à celles retenues pour l'accès précoce et permettent la liquidation des remises dues.
Toutefois, le III de l'article 30 reporte au 1 er janvier 2025 au plus tard l'entrée en vigueur complète des modalités de liquidation des remises prévues aux articles L. 162-16-5-2 et L. 162-16-5-3 du code de la sécurité sociale pour les spécialités dispensées en officine et bénéficiant d'un accès compassionnel. Dans l'attente « d'une remontée d'information qui sera rendue possible par la prescription électronique » 444 ( * ) à cette date, les remises demeurent calculées sur la base d'une fraction du CA annuel réalisé pour la spécialité toutes indications confondues, et non du CA réalisé au titre de l'indication concernée.
b) L'application des remises aux médicaments acquis par l'Agence nationale de santé publique
Le 2° du I de l'article 30 rend les remises prévues dans le cadre des autorisations d'accès dérogatoire applicables aux médicaments acquis par l'Agence nationale de santé publique (ANSP) au titre de l'article L. 1413-4 du code de la santé publique, pour la constitution de stocks stratégiques face aux menaces sanitaires graves ou aux besoins de santé publique non couverts. L'étude d'impact jointe au PLFSS précise qu'il s'agit là « d'assurer l'équité de traitement des produits » 445 ( * ) .
c) L'accélération de la procédure d'accès précoce lorsque l'Agence européenne des médicaments s'est prononcée
Enfin, le II de l'article vise à accélérer, en la simplifiant, la procédure d'autorisation d'accès précoce dans les cas où le comité des médicaments à usage humain (CMUH) de l'Agence européenne des médicaments a rendu un avis positif sur le médicament.
À cette fin, il modifie l'article L. 5121-12 du code de la santé publique pour prévoir que, dans les cas où le médicament concerné ne dispose pas encore d'une AMM dans l'indication considérée mais a en revanche reçu un avis favorable du CMUH :
- l'accès précoce peut être autorisé par la HAS sans avis préalable et conforme de l'ANSM ;
- le protocole d'utilisation thérapeutique et de recueil des données, au respect duquel l'autorisation est subordonnée, est établi par la seule HAS, sans intervention nécessaire de l'ANSM.
B. Réformer les modalités de financement des thérapies innovantes les plus coûteuses
1. Le coût des thérapies innovantes soulève la question de leur mode de financement
Le règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 446 ( * ) distingue quatre types de médicaments de thérapie innovante (MTI) 447 ( * ) :
- les médicaments de thérapie génique (MTG) , soit contenant une substance active qui elle-même contient ou constitue un acide nucléique recombinant administré à des personnes en vue de réguler, réparer, remplacer, ajouter ou supprimer une séquence génétique 448 ( * ) ;
- les médicaments de thérapie cellulaire somatique (MTCS) , qui contiennent ou consistent en des cellules ou des tissus qui ont fait l'objet d'une manipulation substantielle de façon à modifier leurs caractéristiques biologiques, leurs fonctions physiologiques ou leurs propriétés structurelles 449 ( * ) ;
- les produits issus de l'ingénierie tissulaire (PIIT) , contenant des cellules ou tissus issus de l'ingénierie cellulaire ou tissulaire, et capables de régénérer, réparer ou remplacer un tissu humain ;
- les MTI combinés , appartenant à l'un des trois précédents types mais incorporant un ou plusieurs dispositifs médicaux.
Les MTI sont, pour beaucoup, dispensés à l'hôpital et remboursés grâce à la « liste en sus », permettant la prise en charge par l'assurance maladie sur présentation des factures de spécialités pharmaceutiques, dans certaines indications définies, en sus des tarifs d'hospitalisation lorsque ces indications présentent un caractère innovant 450 ( * ) .
Porteurs de bénéfices thérapeutiques parfois très importants, les MTI s'avèrent toutefois très onéreux pour les établissements de santé comme, in fine , pour l'assurance maladie. L'étude d'impact jointe au PLFSS souligne ainsi leur prix croissant, affirmant que « le seuil de deux millions d'euros par patient est désormais franchi » et, au-delà, que « ces traitements sont très consommateurs de ressources hospitalières, car ils exigent un recours à des équipes d'expertise poussée et disposant de plateaux techniques adaptés » 451 ( * ) .
À cet égard, les dépenses de produits de santé au titre de la liste en sus apparaissent particulièrement dynamiques ces dernières années : avant prise en compte des remises et de la clause de sauvegarde, elles ont en moyenne augmenté de 11,9 % par an entre 2019 et 2021, malgré la faible croissance des dispositifs médicaux en raison de la déprogrammation d'opérations durant la crise sanitaire 452 ( * ) . Dans son rapport d'activité 2020, le CEPS estime que les ventes de la liste en sus ont progressé, pour les seuls médicaments, de 20,6 % entre 2019 et 2020. Neuf spécialités innovantes concentreraient 55 % de la dépense totale 453 ( * ) .
Évolution des dépenses brutes de produits de santé de la liste en sus
(en millions d'euros)
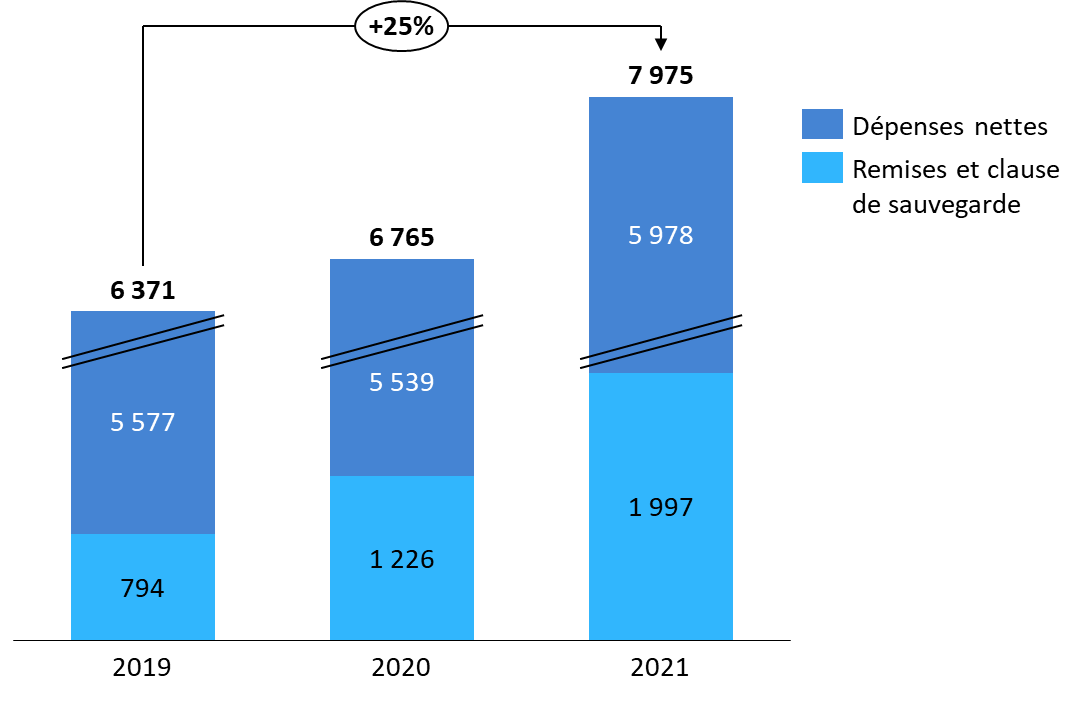
Source : Commission des affaires sociales du Sénat, d'après des données CCSS
2. L'article 30 instaure des modalités de financement spécifiques des MTI les plus coûteux
Afin de ne pas faire porter par les hôpitaux l'avance du coût des médicaments et pour maîtriser le risque financier attaché aux spécialités les plus onéreuses, l'article 30 vise à mettre en place des modalités de financement spécifiques des MTI, permettant de transférer la gestion d'une partie des flux financiers à l'assurance maladie et de conditionner le paiement aux résultats obtenus en vie réelle.
À cette fin, le 3° de l'article insère un nouveau paragraphe à l'article L.162-16-6 du code de la sécurité sociale, fixant les modalités de tarification des médicaments de la liste en sus. Celui-ci prévoit que, lorsque le prix demandé par un industriel pour un MTI inscrit sur la liste en sus est supérieur à un seuil défini par arrêté, le coût du traitement est fixé conventionnellement ou, à défaut, par décision du CEPS.
Un arrêté devra par ailleurs définir un « forfait de thérapie innovante », correspondant au montant maximal que les hôpitaux doivent être amenés à décaisser pour l'acquisition d'un MTI. En conséquence, le tarif de responsabilité sera fixé de telle sorte que le montant correspondant au nombre d'unités de médicament nécessaires multiplié par le tarif de responsabilité soit inférieur au dit forfait. Le prix limite de vente devra être égal au tarif de responsabilité.
Lorsque le coût défini par convention ou décision du CEPS est supérieur à ce montant, son règlement est réalisé par l'assurance maladie, en un ou plusieurs virements annuels successifs. Les modalités de ce paiement échelonné - nombre, montants, conditions et échéances des versements - sont fixées par la convention conclue avec le CEPS ou par sa décision, en tenant compte des données d'efficacité du médicament concerné. Ils sont interrompus en cas d'échec du traitement pour un patient, notamment en cas de décès ou d'administration d'un autre traitement de même visée thérapeutique. Dans ce cas, le coût du traitement, réduit des remises conventionnelles versées, ne peut excéder le coût net des traitements à même visée thérapeutique.
Enfin, l'entreprise assure à sa charge le recueil des données permettant d'évaluer l'efficacité en vie réelle du traitement. À cette fin, les prescripteurs lui transmettent les données de suivi des patients traités.
C. Maîtriser les prix des médicaments
1. La régulation microéconomique du médicament par le CEPS
Trois modes de régulation microéconomique des dépenses de médicament sont principalement exploités par le CEPS.
D'abord, le CEPS fixe, initialement, le prix des médicaments remboursables , en tenant compte principalement de l'amélioration du service médical rendu (ASMR), des prix des médicaments à même visée thérapeutique, des volumes de vente anticipés ainsi que des conditions prévisibles et réelles d'utilisation du médicament. Sont ainsi établis par convention ou, à défaut, décision du CEPS :
- le prix fabricant hors taxes des médicaments vendus en officine 454 ( * ) ;
- le prix de cession au public des produits en rétrocession 455 ( * ) ;
- le prix de responsabilité et le prix limite de vente aux hôpitaux des produits de la liste en sus 456 ( * ) .
Ensuite, le CEPS conduit chaque année un programme de baisses de prix , visant à permettre le respect des objectifs fixés par le Parlement lors du vote de la LFSS. Les prix fixés initialement peuvent être révisés à la baisse, conventionnellement ou, à défaut, par décision du CEPS 457 ( * ) , dans plusieurs circonstances : par exemple en cas de modification significative des données scientifiques et épidémiologiques prises en compte pour la conclusion des conventions, à l'occasion d'un renouvellement d'inscription ou de la commercialisation des premiers génériques d'un groupe inscrit au répertoire. La LFSS pour 2017 458 ( * ) a précisé à l'article L. 162-16-4 du code de la sécurité sociale les critères pouvant motiver une telle baisse : figurent parmi ceux-ci l'ancienneté de l'inscription de la spécialité, le prix net des médicaments à même visée thérapeutique ou encore le prix d'achat constaté de la spécialité par les établissements de santé ou les distributeurs de gros ou de détail. Les baisses de prix ont permis une économie de 1 076 millions d'euros en 2019 459 ( * ) et de 754 millions d'euros en 2020 460 ( * ) .
Enfin, les remises conventionnelles constituent désormais le levier de régulation de la dépense remboursable le plus important. Leur montant a été multiplié par plus de sept en 2012, et s'est établi à 3 406 millions d'euros en 2020, après déduction des avoirs sur remises 461 ( * ) . Trois catégories de remises peuvent être distinguées :
- les remises dites « produits », représentant plus de 90 % du total, amenant les entreprises à reverser à la Cnam tout ou partie du chiffre d'affaires de certaines spécialités remboursables 462 ( * ) ;
- les remises associées aux procédures dérogatoires d'accès : accès précoce, accès compassionnel 463 ( * ) ;
- les remises exonératoires de la clause de sauvegarde 464 ( * ) .
Économies générées par les mesures de régulation du CEPS sur les médicaments remboursables
(en millions d'euros)
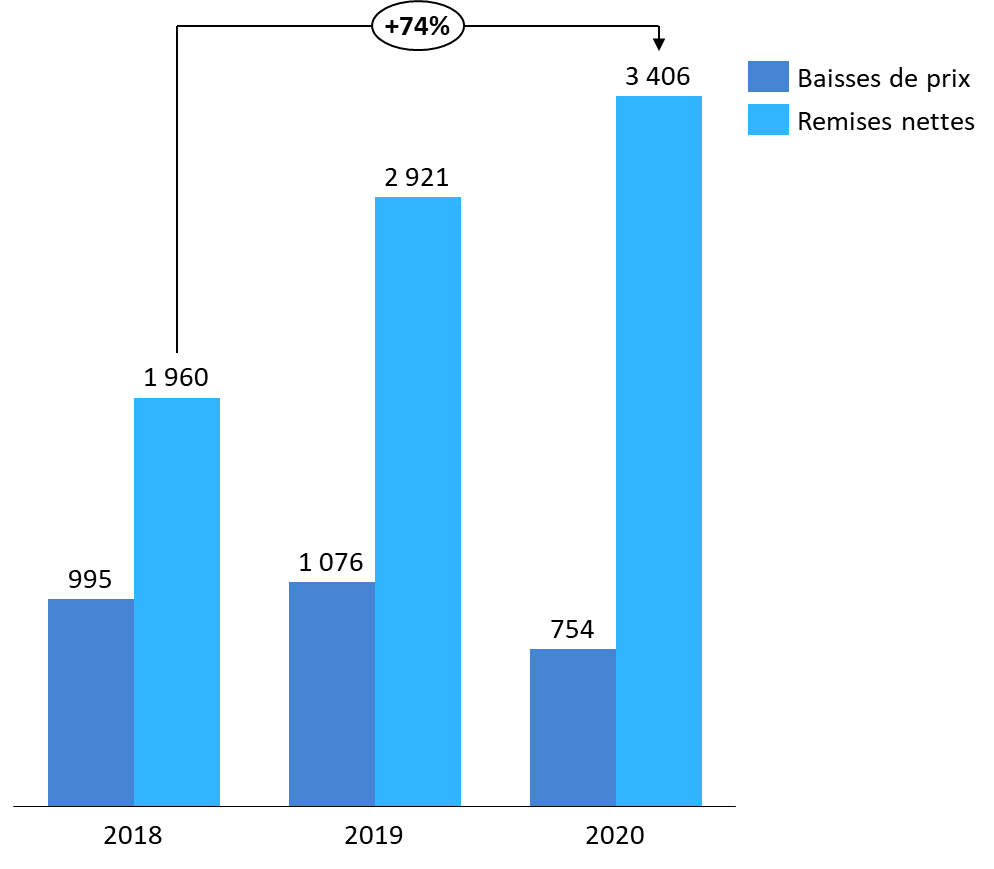
Source : Commission des affaires sociales du Sénat, sur la base des rapports d'activité du CEPS
L'effort de développement des remises produits mené par le CEPS se concentre, pour l'essentiel, sur les thérapies innovantes. Celles-ci sont fixées conventionnellement avec les entreprises. Pour les seules spécialités utilisées en association dans le cadre d'un traitement, ou certaines thérapies géniques ou cellulaires, elles peuvent être imposées unilatéralement par le CEPS 465 ( * ) .
2. L'article 30 vise à renforcer la régulation microéconomique en créant une procédure de référencement périodique et en favorisant le recours aux remises
a) La création d'une procédure de référencement périodique
Afin de renforcer encore la régulation des prix sur les médicaments matures, notamment les génériques, hybrides et biosimilaires, le 4° du I de l'article 30 du PLFSS insère ainsi dans le code de la sécurité sociale un nouvel article L. 162-17-1-3 visant à instituer une procédure dite de « référencement périodique ».
Ces nouvelles dispositions ouvrent la possibilité de subordonner l'inscription sur la liste des médicaments remboursables ou la liste en sus hospitalière à une procédure de référencement visant à sélectionner un ou plusieurs médicaments parmi des spécialités ayant une même visée thérapeutique, sur la base de critères pouvant tenir :
- au volume des médicaments nécessaires pour garantir un approvisionnement suffisant du marché ;
- à l'intérêt des conditions tarifaires proposées ;
- aux objectifs de développement durable et à celui de sécurité d'approvisionnement que garantit l'implantation des sites de production.
Les médicaments ainsi sélectionnés seraient référencés pour une période maximale d'un an, le cas échéant prorogeable de six mois. Les autres médicaments comparables pourraient être, dans la même période, exclus de la prise en charge, sans qu'il soit toutefois possible de placer une entreprise en situation de monopole sur une spécialité.
Enfin, en contrepartie, les entreprises sélectionnées pourraient devoir s'engager à fournir des quantités minimales de médicaments sur le marché français et à garantir une couverture suffisante du territoire pendant toute la durée du référencement. Dans le cas contraire, le Gouvernement pourra :
- mettre fin à la procédure de référencement ou y déroger pour pallier la défaillance constatée ;
- prononcer une pénalité financière à l'encontre de l'entreprise fautive, qui ne peut excéder 10 % de son CA réalisé en France ;
- mettre à sa charge les surcoûts supportés par l'assurance maladie du fait du défaut d'approvisionnement ou de la mauvaise couverture du territoire.
Le système de référencement périodique proposé vise, en stimulant par appel d'offres la compétition entre les entreprises sur des classes de médicaments matures et concurrentielles, à favoriser une baisse des prix. L'étude d'impact jointe au PLFSS estime que la procédure pourrait générer, à terme, près de 100 millions d'euros d'économies en année pleine 466 ( * ) .
b) Favoriser le recours aux remises
Pour maîtriser les dépenses d'assurance maladie et orienter les comportements des entreprises, l'article 30 du PLFSS porte trois mesures visant à favoriser le recours aux remises.
Le 7° du I de l'article , d'abord, étend la faculté du CEPS d'imposer des remises unilatéralement à l'ensemble des médicaments en supprimant, à l'article L. 162-18 du code de la sécurité sociale, les conditions tenant à la nature du médicament ou à son utilisation en association à une autre spécialité. L'étude d'impact jointe au PLFSS précise que cette évolution pourrait permettre d'accélérer les négociations, notamment dans le cas des médicaments innovants pour lesquels les demandes tarifaires peuvent s'avérer très élevées et déconnectées du prix des alternatives remboursées 467 ( * ) .
Par ailleurs, le 8° du I insère dans le code de la sécurité sociale un nouvel article L. 162-18-2 prévoyant l'application obligatoire de remises aux médicaments faisant l'objet d'une demande de remboursement dans un périmètre d'indications thérapeutiques plus restreint que celui de l'AMM délivrée. Ces remises demeureraient dues jusqu'à ce que l'entreprise demande la prise en charge de cette spécialité pour l'ensemble des indications autorisées. Le taux de remise appliqué serait défini selon un barème fixé par arrêté. Pour permettre la liquidation, l'entreprise serait contrainte de déclarer, au plus tard le 15 février n+1, le chiffre d'affaires réalisé pour cette spécialité.
Le IV de l'article laisse aux entreprises exploitant une spécialité inscrite au 1 er janvier 2023 un an pour demander la prise en charge de cette spécialité sur l'ensemble des indications thérapeutiques, en prévoyant l'application des remises obligatoires dans le cas contraire à compter du 1 er janvier 2024.
D'après l'étude d'impact, ces dispositions visent à lutter contre l'« une des stratégies pour obtenir des prix élevés », consistant à revendiquer « un périmètre de remboursement réduit, visant une indication dont la population cible est faible et pour laquelle le traitement a l'effet le plus probant », alors même que l'AMM obtenue couvre un périmètre plus large 468 ( * ) .
Enfin, le 8° du I insère également dans le code de la sécurité sociale un nouvel article L. 162-18-3 permettant de sanctionner les entreprises ne se conformant pas aux obligations déclaratives destinées à permettre la liquidation des remises en majorant de 2 % ces dernières par semaine de retard. Un décret déterminera les conditions d'application de ces majorations, afin qu'une part minimale du chiffre d'affaires correspondant aux spécialités concernées ne soit pas soumise à reversement.
D. Garantir l'approvisionnement en médicaments d'intérêt thérapeutique majeur
1. L'approvisionnement en médicaments : un enjeu important
Du fait de leur importance dans le traitement des patients, des dispositifs de lutte contre les ruptures d'approvisionnement des médicaments d'intérêt thérapeutique majeur (MITM) ont progressivement été renforcés ces dernières années.
Série 1
Depuis la loi de 2016 de modernisation de notre système de santé 469 ( * ) , les MITM sont définis par le code de la santé publique comme ceux « pour lesquels une interruption de traitement est susceptible de mettre en jeu le pronostic vital des patients à court ou moyen terme, ou représente une perte de chance importante pour les patients au regard de la gravité ou du potentiel évolutif de la maladie » 470 ( * ) .
Un chapitre du code de la santé publique relatif à la « lutte contre les ruptures d'approvisionnement de médicaments » fait désormais obligation aux exploitants de MITM :
- de constituer un stock de sécurité destiné au marché national 471 ( * ) , défini depuis le 1 er septembre 2021 comme correspondant à au moins deux mois de couverture des besoins 472 ( * ) ;
- d'élaborer des plans de gestion des pénuries dont l'objet est de prévenir et pallier toute rupture 473 ( * ) ;
- d'informer l'ANSM, dès qu'ils en ont connaissance, de tout risque de rupture ou de toute rupture 474 ( * ) .
Ces mesures n'ont toutefois pas permis de réduire les tensions dans l'approvisionnement en MITM ces dernières années. Au contraire, le nombre de signalements transmis à l'ANSM a continué d'augmenter, pour s'établir à 2 160 en 2021.
Nombre de signalements de rupture et de risques de rupture transmis à l'ANSM
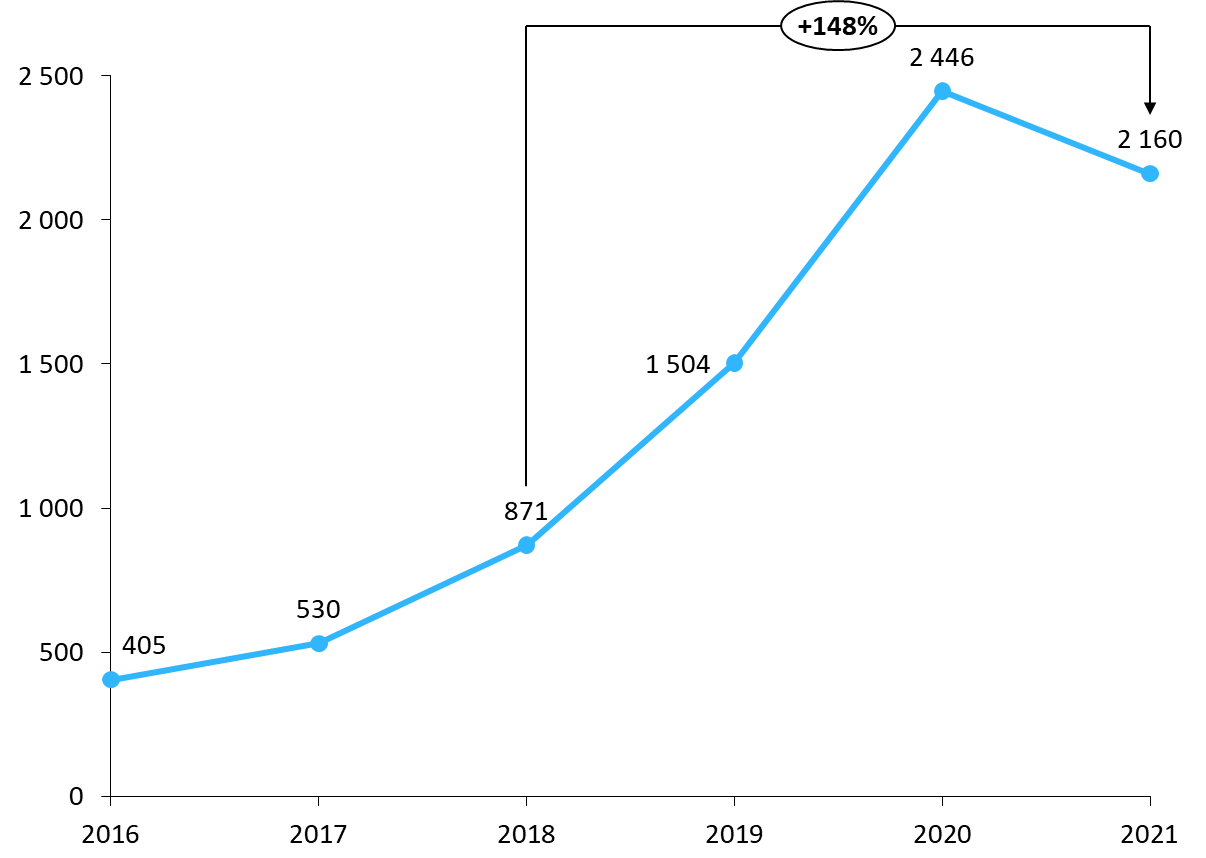
Source : Commission des affaires sociales, d'après les données de l'ANSM
2. L'article 30 vise à inciter les professionnels à maintenir l'accès à leurs MITM anciens
Relevant, dans l'étude d'impact, des « choix stratégiques de certains laboratoires pharmaceutiques de concentrer leur activité sur de nouveaux produits à forte marge et donc d'abandonner l'exploitation d'autres produits matures moins rentables », le Gouvernement souhaite inciter les professionnels à maintenir l'accès à leurs MITM anciens lorsque de nouvelles spécialités, plus rémunératrices, sont autorisées.
À cette fin, le 6° du I de l'article 30 insère dans le code de la santé publique un article L. 162-17-4-4 prévoyant que, dans le cadre d'une première demande d'inscription sur la liste des médicaments remboursables, la liste de rétrocession ou la liste des spécialités agréées pour les hôpitaux, d'une spécialité dont l'ASMR appréciée par la HAS est au moins d'un niveau fixé par décret, l'entreprise s'engage à garantir l'approvisionnement des MITM déjà inscrits par convention avec le CEPS.
En cas de manquement, et après que l'entreprise a été mise en mesure de présenter ses observations, le CEPS pourra prononcer une pénalité financière plafonnée à 10 % du chiffre d'affaires réalisé en France par l'entreprise au titre du médicament considéré. La pénalité sera reconductible chaque année en cas de persistance du manquement. Son produit sera affecté à la Cnam 475 ( * ) .
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Dans le texte considéré comme adopté par l'Assemblée nationale suite à l'engagement, par le Gouvernement, de sa responsabilité, l'article 30 est substantiellement modifié.
Un premier amendement complète le 3° du I de l'article pour préciser que, dans le cadre des nouvelles modalités de financement des MTI les plus onéreux, la prise en charge par l'assurance maladie s'effectue :
- d'une part, à hauteur du tarif de responsabilité, par le remboursement de l'établissement de santé ;
- d'autre part, lorsque le coût de traitement est supérieur au tarif de responsabilité multiplié par le nombre d'unités, par un ou plusieurs versements directs à l'entreprise.
Surtout, outre plusieurs amendements rédactionnels, le Gouvernement a accepté deux amendements visant à revenir sur deux dispositifs que l'article 30 visait à mettre en place.
D'une part, un amendement de Philippe Vigier et plusieurs de ses collègues supprime le 4° du I de l'article instaurant la procédure de référencement périodique. En substitution, l'amendement ajoute un III bis prévoyant que le Gouvernement remet au Parlement, avant le 1 er juillet 2023, un rapport évaluant l'intérêt, la faisabilité et les potentielles limites d'un tel dispositif, notamment à l'appui d'exemples étrangers, « en vue d'en proposer une disposition dans un prochain [PLFSS] ».
D'autre part, un amendement de Thomas Mesnier supprime le 6° du I qui visait à inciter les entreprises à maintenir l'accès aux MITM anciens malgré l'introduction de nouvelles spécialités innovantes et rémunératrices.
L'Assemblée nationale est considérée comme ayant adopté cet article ainsi modifié.
III - La position de la commission : des mesures éparses et insuffisamment préparées
L'article 30 contient de nombreuses mesures éparses sur le médicament , visant tour à tour à en améliorer l'accès ou l'approvisionnement, à mieux réguler les prix et les dépenses d'assurance maladie ou à redéfinir les modalités de financement et de paiement, dont la cohérence n'est pas toujours apparue évidente à la commission. Ce « pointillisme » législatif nuit à la lisibilité du texte.
Surtout, ces dispositions sont apparues, au fil de leur examen par l'Assemblée nationale, insuffisamment préparées avec l'ensemble des parties prenantes : les inquiétudes exprimées par les industriels comme par les pharmaciens ont été nombreuses et le Gouvernement a accepté de revenir sur plusieurs des mesures phares, pourtant présentées dans l'étude d'impact comme des sources d'économie et d'encadrement indispensables.
Certaines mesures portées par le texte favorisent un meilleur accès aux médicaments, notamment innovants , et ont, à ce titre, reçu l'assentiment de la commission. Il en va ainsi des améliorations apportées aux dispositifs d'accès dérogatoire aux soins, et notamment de l'accélération de la procédure d'accès précoce lorsque l'avis favorable du CMUH suffit à présumer de la sécurité et de l'efficacité d'un traitement innovant.
De la même manière, le nouveau dispositif de financement des médicaments de thérapie innovante , fondé sur des paiements échelonnés de l'assurance maladie conditionnés à l'obtention de résultats probants en vie réelle, permettra de réduire l'avance de trésorerie consentie par les hôpitaux et de maîtriser le risque financier attaché à ces innovations. Si cette mesure a été approuvée par les représentants des établissements de santé comme par ceux des entreprises entendus, plusieurs points de vigilance ont toutefois été signalés à la rapporteure :
- les demandes de remboursement et les modalités de recueil des données d'efficacité en vie réelle des MTI ne devront pas s'avérer coûteux, en gestion, pour les établissements de santé ou devront être, dans le cas contraire, justement valorisés ;
- les critères devant permettre d'évaluer l'efficacité en vie réelle des médicaments concernés devront faire l'objet d'un consensus scientifique ;
- enfin, le Gouvernement devra veiller à ce que les tarifs de responsabilité, définis pour ne pas excéder le forfait correspondant au montant maximal pouvant être décaissé par les hôpitaux, ne constituent pas à l'international un signal erroné sur le prix de ces traitements : il s'agit là d'une crainte importante, et légitime, des industriels concernés.
La commission a toutefois jugé inopportune l'extension à l'ensemble des médicaments de la faculté du CEPS d'imposer des remises unilatéralement . Celle-ci risquerait d'adresser un mauvais signal aux entreprises du secteur et d'entraver la politique de régulation menée par le CEPS, privilégiant jusque-là la négociation de conventions avec les exploitants. Interrogé par la rapporteure, le CEPS a d'ailleurs confirmé qu'il n'avait jamais fait usage de cette faculté, pourtant autorisée pour les médicaments en association ou certains traitements innovants. Jugeant l'utilité de ces dispositions très incertaine et tenant compte des effets contreproductifs qu'elles risqueraient d'engendrer dans le climat et l'équilibre des négociations, la commission a adopté à l'initiative de la rapporteure un amendement n° 78 visant à supprimer le 7° du I de l'article 30 étendant à l'ensemble des médicaments les remises unilatérales.
Enfin, la commission a jugé favorablement la suppression de la procédure de référencement périodique, insuffisamment préparée et contre laquelle de nombreux acteurs du secteur s'étaient élevés. Dans le cas où il souhaiterait proposer à nouveau cette mesure dans un prochain texte, le Gouvernement devra tenir compte des inquiétudes des pharmaciens comme des industriels, qui ont notamment souligné que cette procédure méconnaissait les conditions de production des médicaments.
Afin de ne pas écourter cette concertation nécessaire, et conformément à sa position habituelle sur les demandes de rapport, la commission a adopté un amendement n° 79 de sa rapporteure visant à supprimer le nouveau III bis. de l'article 30 demandant au Gouvernement de remettre au Parlement un rapport relatif au référencement périodique pour le 1 er juillet 2023. Si, comme semblent l'indiquer ces dispositions, les réflexions à venir devaient conduire le Gouvernement à proposer à nouveau cette mesure dans un prochain texte, l'étude d'impact jointe à celui-ci devrait décrire précisément les conséquences économiques, financières et sociales attendues d'un dispositif aussi structurant.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article 30 bis
(nouveau)
Procédure d'inscription des spécialités sur
la liste de prise en charge
Cet article, inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité en application du troisième alinéa de l'article 49 de la Constitution, vise à clarifier la procédure d'admission des spécialités de la liste dite « de rétrocession » à la prise en charge par l'assurance maladie.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
A. Le droit existant : la liste « en rétrocession »
Au sein des établissements de santé, les pharmacies à usage intérieur (PUI) répondent aux besoins pharmaceutiques des personnes prises en charge par l'établissement ou le groupement hospitalier de territoire (GHT) dont elles relèvent. À cet effet, elles assurent notamment la gestion, l'approvisionnement, la préparation et la dispensation des médicaments ou dispositifs médicaux stériles 476 ( * ) .
Toutefois et par dérogation, les PUI sont autorisées à vendre directement au public certains médicaments figurant sur une liste, dite « liste en rétrocession » , établie par le directeur général de l'Agence nationale de sécurité du médicament (ANSM) depuis l'entrée en vigueur de la loi d'accélération et de simplification de l'action publique 477 ( * ) .
L'inscription d'un médicament sur cette liste doit être justifiée par des raisons de santé publique, l'intérêt des patients ou, le cas échéant, résulter d'une demande du Gouvernement 478 ( * ) . Les médicaments faisant l'objet d'une autorisation d'accès précoce ou d'une autorisation de prescription compassionnelle sont réputés y figurer 479 ( * ) .
L'article L. 162-17 du code de la sécurité sociale dispose que les médicaments de la liste en rétrocession sont pris en charge par l'assurance maladie , pour les indications thérapeutiques y ouvrant droit, « lorsqu'ils sont délivrés par une pharmacie à usage intérieur d'un établissement de santé dûment autorisé ».
B. Le droit proposé : une clarification des modalités de prise en charge de ces spécialités
Le présent article, issu d'un amendement du Gouvernement conservé dans le texte sur lequel ce dernier a engagé sa responsabilité, vise à clarifier les conditions de prise en charge des médicaments de la liste en rétrocession.
À cette fin, l' article 30 bis complète l'article L. 162-17 du code de la sécurité sociale pour prévoir que les médicaments inscrits sur la liste en rétrocession délivrés par une PUI dûment autorisée doivent, pour être pris en charge, figurer sur une liste établie dans des conditions fixées par décret en Conseil d'État.
Cette seconde liste précisera les indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie.
II - La position de la commission
Cet article vise à clarifier les dispositions issues de la loi d'accélération et de simplification de l'action publique, en précisant que la prise en charge des médicaments de la liste en rétrocession est conditionnée à leur inscription sur une liste de prise en charge.
Ces modalités d'autorisation de prise en charge sont identiques à celles prévues pour les médicaments dispensés en ville 480 ( * ) .
La commission vous demande d'adopter cet article sans modification.
Article 30 ter
(nouveau)
Prolongation de l'expérimentation du cannabis
thérapeutique
Cet article, inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité en application du troisième alinéa de l'article 49 de la Constitution, prolonge d'un an l'expérimentation du cannabis thérapeutique, lancée par la LFSS pour 2020.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé : une expérimentation prolongée d'un an
L'article 30 ter fait passer la durée de l'expérimentation du cannabis à usage médical, qui était fixée à deux ans par la loi de financement de la sécurité sociale pour 2020 481 ( * ) , à trois ans.
II - La position de la commission : adoption conforme
L'évaluation du dispositif prévu par la LFSS pour 2020 avait pour objectif principal d'estimer la faisabilité du circuit de mise à disposition du cannabis médical pour les patients aux différentes étapes du parcours de prise en charge, de la prescription à la dispensation, et pour objectif secondaire d'assurer le recueil des premières données françaises sur l'efficacité de l'utilisation du cannabis dans un cadre médical, ainsi que des données associées à la consommation de soins afin de pouvoir se positionner sur le futur cadre de prise en charge.
Les outils d'évaluation étaient les suivants :
- un comité scientifique temporaire composé par l'ANSM, créé pour suivre l'expérimentation dans tous ses paramètres : nombre de patients, modalités de suivi et de dispensation, etc . ;
- l'évaluation des données sur la faisabilité du circuit de distribution et les premières données d'efficacité, menée par la société Iqvia à partir du registre national de suivi ReCann mis en place par l'ANSM ;
- une étude des effets indésirables, au moyen d'une extraction des données de la base nationale de pharmacovigilance ;
- une enquête auprès des patients sur leur parcours et leurs perceptions, réalisée par ViaVoice ;
- une étude visant à évaluer l'impact chez les patients de l'utilisation du cannabis à visée médicale, réalisée par un centre de recherche de l'Inserm à partir des données issues du système national des données de santé (SNDS).
Au 31 mars 2022, 1 450 patients avaient été inclus dans l'expérimentation, et 69 % l'étaient toujours.
Les données analysées par Iqvia ont permis de conclure à la faisabilité du circuit de mise à disposition du cannabis médical « pour la majorité des étapes » : le parcours patient, la réalisation de la titration et le circuit logistique du produit sont satisfaisants, mais le relais de la prise en charge en ville et le délai de dispensation du traitement par la pharmacie au patient « ne permettent pas de conclure concernant leur faisabilité ». L'étude invoque pour l'expliquer la « nécessaire appropriation par les professionnels » de la nouvelle thérapeutique.
Les données de pharmacovigilance et d'addictovigilance de l'expérimentation du cannabis médical montrent un profil de sécurité conforme à ses caractéristiques pharmacologiques et aux données disponibles . Peu d'évènements indésirables graves ont été signalés. Sur la période étudiée, aucun cas d'abus et de dépendance avec le cannabis médical n'est rapporté dans l'expérimentation française, mais « les données d'addictovigilance sont très limitées et les antécédents addictologiques des patients, notamment les consommations de cannabis récréatif et de CBD non médical, sont très peu renseignées ». Ces informations ne sont en effet pas recueillies dans le registre Recann, la Cnil ayant estimé que cette donnée, relative à une substance illicite dont la consommation est interdite en France, était trop sensible. Le rapport précise à cet égard « qu'un travail avec le réseau des centres antipoison français a été mis en place afin d'étudier le risque en cas d'intoxication ».
L'enquête réalisée auprès des patients, menée du 1 er au 22 juillet dernier sur un échantillon de 725 personnes, confirme la faible participation des prescripteurs en ville vis-à-vis des structures de référence. Cependant, les patients interrogés sont globalement satisfaits de la facilité de prise en charge et de contact des centres de référence. Une majorité des répondants ont perçu des effets bénéfiques liés au traitement, qu'ils soient physiques ou psychologiques, ou une amélioration de leur qualité de vie.
L'étude sur l'utilisation concomitante des traitements et autres thérapies chez les patients n'a toutefois pu être conduite , les données du SNDS n'ayant pu être mises à disposition des équipes chargées de leur analyse « en raison d'un programme de travail extrêmement chargé sur de nombreuses autres priorités ministérielles ».
D'importantes disparités régionales concernant l'accès au traitement ont enfin été mises en exergue, notamment en raison d'une répartition non homogène sur le territoire des médecins autorisés et souhaitant prescrire les médicaments à base de cannabis. Une faible proportion de patients, moins d'un sur dix, ont ainsi pu bénéficier du relais hôpital-ville.
L'évaluation communiquée au Sénat le 18 octobre dernier fait en conclusion apparaître qu'une prolongation de l'expérimentation :
- permettrait la réalisation de l'étude tirée des données du SNDS précitée ;
- « pourrait permettre d'offrir un accès de l'offre plus équilibré sur l'ensemble des régions » s'il était associé à « un plan d'actions ambitieux » pour mobiliser davantage les médecins généralistes ;
- « serait de nature à atteindre la cible initialement fixée à 3 000 patients inclus et de disposer d'une file active plus importante ».
Le rapport d'évaluation estime en définitive que la prolongation « permettrait de confirmer la sécurité sanitaire des médicaments à base de cannabis, et d'accompagner la bonne compréhension et l'acceptabilité sociétale d'une entrée en droit commun du dispositif ».
La rapporteure prend note de ces premiers éléments d'évaluation. Elle estime que, s'il importe certes de ne pas laisser sans solution thérapeutique les patients inclus dans les premiers mois de l'expérimentation et qui semblent, d'après l'enquête, bien supporter le traitement, il importe tout autant de disposer, sur l'usage thérapeutique d'un produit aux effets ambivalents et à l'image sociale complexe 482 ( * ) , d'éléments d'aide à la décision fiables et aussi complets que possible.
La commission vous demande d'adopter cet article sans modification.
Article 31
Garantir
l'accès des patients aux dispositifs médicaux, produits et
prestations et l'efficience de leur prise en charge
Cet article vise :
- à améliorer la tarification des dispositifs médicaux et des prestations associées en distinguant mieux la valorisation du produit, de la marge de distribution et des prestations associées et en réformant, pour certains de ces éléments, les modalités de fixation du prix ;
- à renforcer la régulation des inscriptions en veillant à ce que leur champ couvre l'ensemble du périmètre du marquage CE, en améliorant les contrôles des lignes génériques et en favorisant les études post-inscription ;
- à compléter, à la marge, les régimes de prise en charge des dispositifs innovants comme de la télésurveillance.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
A. Refonder la tarification des produits et prestations de la LPPR
La notion de « dispositif médical » couvre un ensemble extrêmement vaste de produits. Le règlement européen 2017/745 relatif aux dispositifs médicaux 483 ( * ) la définit comme « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme » à des fins médicales. Elle comprend aussi bien des produits consommables (pansements, préservatifs, etc .), des implants (prothèses cardiaques ou de hanche, etc .), des équipements (lits médicaux, fauteuils roulants, IRM, scanners, etc .), que des orthèses et prothèses externes (optique médicale, appareils correcteurs de surdité, etc .).
Les dispositifs médicaux peuvent être commercialisés après obtention d'une marque dite « CE » , attestant de leur conformité aux exigences de sécurité et de performance européennes 484 ( * ) .
Leur prise en charge par l'assurance maladie, en ville ou sur la liste en sus hospitalière, est conditionnée à leur inscription sur la liste des produits et prestations remboursables (LPPR) , après avis de la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) de la Haute Autorité de santé (HAS) 485 ( * ) .
Remboursements de la LPPR, en ville et en 2018
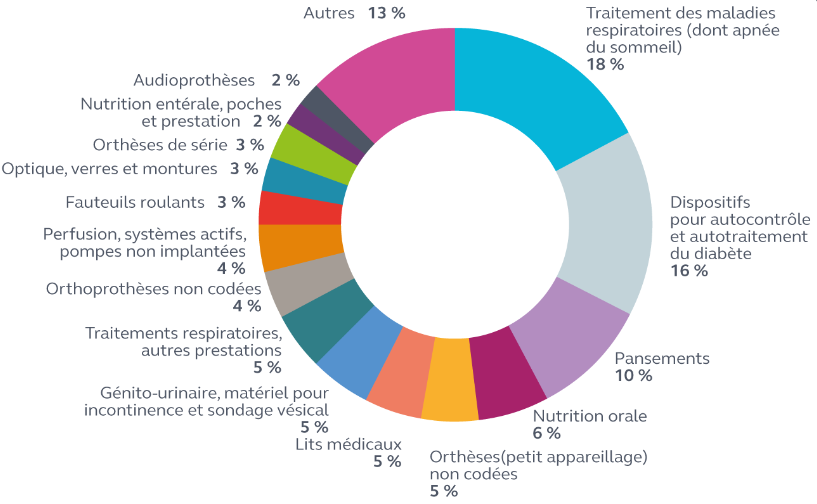
Source : Cour des comptes, Rapport sur l'application des LFSS , octobre 2020
L'article 31 du PLFSS vise à améliorer les modalités de fixation des prix des produits et prestations de la LPPR en :
- distinguant mieux, en son sein, les dispositifs médicaux des marges de distribution et des prestations qui y sont associées ;
- fixant par voie réglementaire la rémunération des distributeurs ;
- contraignant les exploitants à déclarer le prix d'acquisition des dispositifs médicaux qu'ils ne fabriquent pas ;
- précisant, à la marge, le régime juridique des conventions conclues avec le comité économique des produits de santé (CEPS).
1. Dissocier la tarification du produit, de la marge de distribution et de la prestation
a) Les modalités de tarification actuelles
Aux termes de l'article L. 165-1 du code de la sécurité sociale, la LPPR rassemble les dispositifs médicaux à usage individuel, les tissus et cellules issus du corps humain et leurs dérivés, les produits de santé autres que les médicaments et les prestations de services et d'adaptation associées.
Le tarif de responsabilité de ces produits et prestations définit l'assiette de leur prise en charge par l'assurance maladie. Il est établi par convention entre l'exploitant ou le distributeur et le CEPS ou, à défaut, par décision unilatérale de ce dernier. Comme pour le médicament, la fixation de ce tarif tient compte principalement de l'amélioration éventuelle du service attendu ainsi que, le cas échéant, des tarifs des produits ou prestations comparables, des volumes de vente prévus ou constatés, des montants remboursés par l'assurance maladie et des conditions prévisibles et réelles d'utilisation 486 ( * ) .
La distribution des dispositifs médicaux fait intervenir une grande diversité d'acteurs - grossistes, magasins spécialisés, pharmacies d'officine, grande distribution, prestataires de services et distributeurs de matériel (PSDM), professions de santé spécialisées (opticiens, orthoprothésistes...) -, qui peuvent réaliser des prestations associées au dispositif, en fournissant par exemple du matériel et des consommables, une formation à l'utilisation, ou en proposant un suivi du patient.
Or la tarification des produits inscrits à la LPPR inclut aujourd'hui, le plus souvent, la rémunération de l'exploitant, des distributeurs et, le cas échéant, des services associés sans qu'il soit possible de distinguer leurs valorisations respectives .
L'étude d'impact jointe au PLFSS souligne, à cet égard, « l'incertitude actuelle sur la part relative de la valeur attribuée au dispositif médical, celle attribuée à sa distribution voire celle relative à la prestation associée à la dispensation » et estime qu'« il en résulte bien souvent une décorrélation entre cette répartition de la valeur et l'apport respectif du produit, de sa distribution et de l'éventuelle prestation » 487 ( * ) .
b) L'article 31 vise à dissocier le tarif des produits de celui des prestations
Afin de simplifier les modalités de tarification des dispositifs médicaux, et pour valoriser plus justement l'activité de chacun des intervenants, l'article 31 vise à dissocier le tarif des produits des marges de distribution et des éventuelles prestations associées.
À cette fin, le 6° du I de l'article modifie l'article L. 165-1 du code de la sécurité sociale pour prévoir que l'inscription sur la LPPR d'une prestation de service et d'adaptation associée à un produit se fera désormais de manière distincte de l'inscription du produit lui-même. En conséquence, la CNEDiMTS émettra, le cas échéant, un avis distinct sur la prestation de service et d'adaptation associée.
En conséquence, le 10° du I modifie l'article L. 165-2 du même code relatif à la fixation, par le CEPS, des tarifs de responsabilité. Désormais, pour les dispositifs inscrits sur la LPPR sur une ligne générique, le CEPS serait chargé de définir des tarifs de responsabilité distincts pour le produit, d'une part, et pour les prestations associées, d'autre part. Pour les dispositifs princeps inscrits sur la LPPR sous forme de nom de marque ou de nom commercial, le CEPS définirait un tarif de responsabilité couvrant le seul produit. Les éventuelles baisses de tarif initiées par le CEPS sont, logiquement, dissociées de la même manière.
Enfin, le 11° du I modifie l'article L. 165-3 du code de la sécurité sociale, relatif aux conditions de négociation des conventions, dans le même sens. Celui-ci prévoirait désormais :
- pour les produits princeps , que la convention est établie entre l'exploitant du produit concerné et le CEPS, et non plus entre l'exploitant ou le distributeur au détail ;
- pour les produits génériques, que les conventions relatives auxdits produits seront établies séparément de celles relatives aux prestations associées, le CEPS négociant chacune d'entre elles avec les exploitants ou distributeurs concernés, le cas échéant regroupés en organisations.
Le III de l'article, enfin, prévoit que ces dispositions entrent en vigueur dans des conditions et à des dates fixées par décret, au plus tard le 31 décembre 2025, en fonction des catégories de produits ou prestations concernées.
2. Fixer par voie réglementaire la rémunération des distributeurs
a) La rémunération actuelle des distributeurs
Les marges des distributeurs de dispositifs médicaux ne sont pas régulées comme celles des distributeurs de médicaments . Pour ces derniers, le code de la sécurité sociale prévoit que « les ministres chargés de l'économie, de la santé et de la sécurité sociale, peuvent fixer par décision les prix et les marges des produits » 488 ( * ) . En application de ces dispositions, un arrêté fixe des taux limites de marge, selon un barème dégressif fondé sur le prix fabricant hors taxes du médicament 489 ( * ) .
À l'inverse, la rémunération des distributeurs est, pour les dispositifs médicaux, aujourd'hui incluse dans la tarification globale du produit. Ce n'est qu'indirectement que le CEPS peut assurer une marge minimale au distributeur, et maîtriser le reste à charge des assurés, par la définition :
- d'un prix de cession maximal auquel le produit peut être vendu au distributeur de détail 490 ( * ) ;
- d'un prix maximal de vente aux assurés , que le comité est invité à définir chaque fois que « les tarifs de remboursement sont cohérents avec le niveau de prix réel des produits concernés » 491 ( * ) .
b) L'article 31 vise à permettre au pouvoir réglementaire de fixer les marges des distributeurs de dispositifs médicaux
Sur le modèle des dispositions applicables aux médicaments, l'article 31 permet la fixation par le pouvoir réglementaire des marges applicables aux distributeurs de dispositifs médicaux.
Pour ce faire, le 12° du I de l'article insère un nouvel article L. 165-3-4 dans le code de la sécurité sociale qui prévoit :
- que les ministres chargés de l'économie, de la santé et de la sécurité sociale fixeront désormais les marges de distribution des produits inscrits sur la LPPR, en tenant compte de l'évolution des charges, des revenus et du volume d'activité des praticiens ou entreprises concernées ;
- que les remises, ristournes et avantages commerciaux et financiers consentis par les fournisseurs aux distributeurs au détail ne pourront excéder, par année civile et par ligne de produits, un seuil fixé par arrêté dans la limite de 50 % du prix fabricant hors taxes.
Tirant les conséquences de ce nouveau mode de régulation des marges, les 4°, 10° et 11° du I modifient respectivement les articles L. 162-38, L. 165-2 et L. 165-3 du même code pour :
- supprimer la faculté du CEPS, devenue sans objet, de fixer un prix de cession maximal ;
- prévoir que les tarifs de responsabilité et le prix des produits et des prestations inscrits sur la LPPR comprennent les marges prévues, le cas échéant, par arrêté des ministres.
Les modalités d'entrée en vigueur fixées par le III de l'article 31 s'appliquent également à ces dispositions.
3. Contraindre l'exploitant à déclarer le prix d'achat
a) L'exploitant des dispositifs médicaux n'est pas nécessairement le fabricant
L'article L. 165-1-1-1 du code de la sécurité sociale définit l'exploitant d'un produit de santé autre qu'un médicament comme « le fabricant ou son mandataire » ou, à défaut, « le ou les distributeurs qui se fournissent directement auprès du fabricant ou de son mandataire » ou, à défaut encore, « tout distributeur intervenant sur le marché français, à condition que pour chaque produit commercialisé, ce distributeur ne se fournisse pas auprès d'un exploitant [...] ni ne fournisse un autre exploitant ».
Pour améliorer la transparence du secteur , la LFSS pour 2020 a déjà prévu que, lorsque l'exploitant n'est pas le fabricant du produit, il est tenu de déclarer l'identité du fabricant et toute information permettant l'identification certaine du produit 492 ( * ) . Par ailleurs, tout exploitant ou fournisseur de distributeur au détail de produits inscrits sur la LPPR est tenu de déclarer annuellement le prix auquel il a vendu ses produits et prestations au CEPS, ce dernier pouvant sanctionner d'une pénalité toute absence de déclaration ou déclaration manifestement inexacte 493 ( * ) .
En revanche, rien n'oblige l'exploitant non fabricant à déclarer le prix auquel il acquiert, lui-même, les produits auprès du fabricant ou d'un grossiste.
b) L'article 31 vise à contraindre l'exploitant non fabricant à déclarer le prix d'achat des dispositifs médicaux
Pour améliorer la transparence du partage de la valeur entre exploitant et fabricant, le 7° du I de l'article 31 complète l'article L. 165-1-1-1 du code de la sécurité sociale par six alinéas prévoyant :
- que l'exploitant non fabricant est tenu de déclarer auprès des ministres de la santé et de la sécurité sociale le prix auquel il a acheté le produit auprès de son fournisseur, déduction faite des différentes remises ou taxes en vigueur ;
- que lorsque la déclaration n'a pas été effectuée dans les délais et formes précisées par voie réglementaire ou lorsqu'elle s'avère manifestement inexacte, les ministres peuvent prononcer, après que celui-ci a été mis en mesure de présenter ses observations, une pénalité financière à la charge de l'exploitant qui ne peut être supérieure à 5 % du chiffre d'affaires réalisé en France par l'exploitant au titre du dernier exercice clos ;
- que cette pénalité est recouvrée par les Urssaf et affectée à la Cnam.
4. Préciser le régime juridique des conventions conclues avec le CEPS
a) La régulation microéconomique des dispositifs médicaux par voie conventionnelle
Les conventions constituent l'outil privilégié de régulation, par le CEPS, des dépenses d'assurance maladie relatives aux dispositifs médicaux. Ce n'est qu'à défaut d'accord que le CEPS décide unilatéralement.
Les conventions sont chargées, d'une part, de fixer le tarif de responsabilité des dispositifs médicaux, qui peut être révisé à la baisse , par exemple lorsque l'ancienneté de l'inscription, les tarifs des produits et prestations comparables, les volumes de vente prévus ou constatés le justifient 494 ( * ) . Elles prévoient, d'autre part, les remises que les entreprises s'engagent à reverser à la Cnam sur tout ou partie du chiffre d'affaires réalisé en France sur les produits ou prestations de la LPPR 495 ( * ) .
Si les économies permises par ces dispositifs de régulation sont importantes, elles ont rarement atteint ces dernières années les objectifs de baisse des prix fixés en LFSS pour les dispositifs médicaux.
Objectifs d'économies et économies réalisées par baisses de prix sur les dispositifs médicaux
(en millions d'euros)
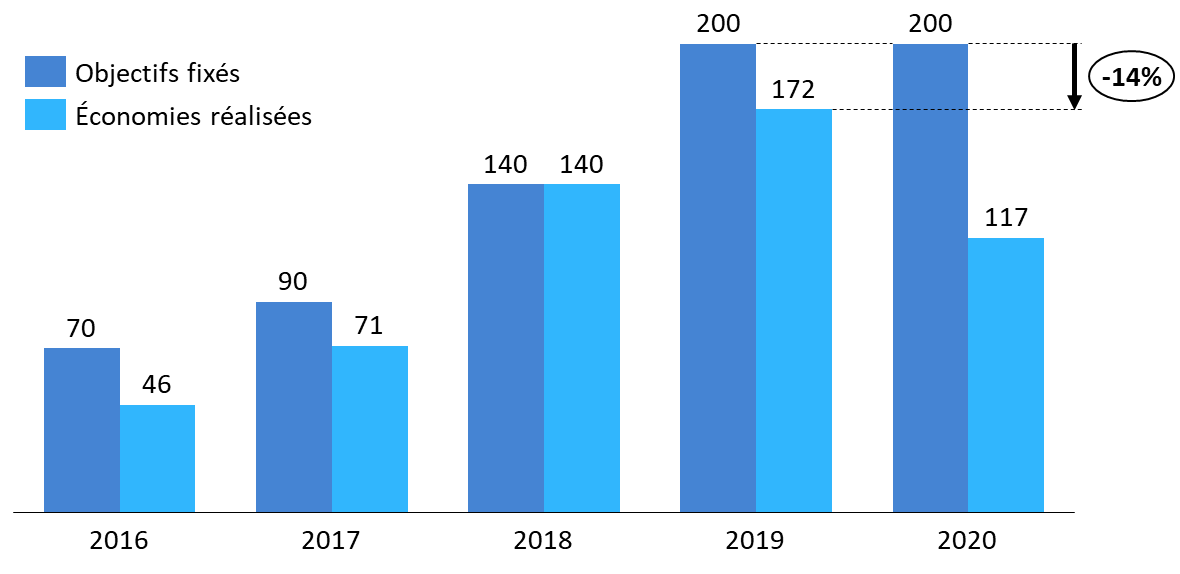
Source : Commission des affaires sociales, données Cour des comptes et rapport annuel 2020 du CEPS
Les conventions conclues par le CEPS avec les entreprises du dispositif médical sont encadrées par l'article L. 165-4-1 du code de la sécurité sociale, lequel renvoie à l'article L. 162-17-4 relatif aux conventions conclues avec les entreprises du médicament. L'étude d'impact jointe au PLFSS souligne que ces dispositions doivent être adaptées aux spécificités de la LPPR, notamment pour prévoir la conduite désormais nécessaire de négociations séparées concernant les produits et les prestations.
b) L'article 31 vise à sécuriser le régime juridique des conventions
Pour mieux définir le régime applicable aux conventions conclues entre le CEPS et les exploitants et distributeurs de produits ou prestations de la LPPR, le 14° de l'article 31 du PLFSS insère au début de l'article L. 165-4-1 du code de la sécurité sociale huit alinéas prévoyant :
- qu'en application des orientations qu'il reçoit chaque année des ministres compétents, le CEPS conclut des conventions relatives aux produits et aux prestations de la LPPR avec les entreprises qui les exploitent et les distribuent ou, en cas d'inscription générique, avec les organisations les regroupant ;
- que les conventions déterminent notamment les remises consenties par les signataires, les conditions et modalités de mise en oeuvre des études postérieures à l'inscription sur la LPPR et les dispositions applicables en cas de non-respect de ces engagements ;
- que, lorsque les conventions conclues ne sont plus compatibles avec les orientations ministérielles, le respect de l'Ondam ou l'évolution des données scientifiques et épidémiologiques, le CEPS peut demander aux signataires de conclure un avenant permettant d'adapter la convention à la situation ou, en cas de refus de ces derniers, résilier la convention ou certaines de ses dispositions et fixer par décision unilatérale le prix des produits et des prestations.
Les modalités d'application de ces dispositions, et notamment les conditions de révision et de résiliation des conventions, doivent être précisées par décret en Conseil d'État.
B. Renforcer la régulation des inscriptions
L'article 31 du PLFSS vise également à mieux réguler les inscriptions sur la LPPR en incitant l'exploitant à demander l'inscription sur l'ensemble des indications autorisées par son marquage CE, en améliorant les contrôles sur les dispositifs inscrits sur ligne générique et en renforçant les évaluations ex post .
1. Inciter l'exploitant à demander l'inscription sur l'ensemble des indications autorisées
a) L'exploitant peut être incité à restreindre sa demande d'inscription à une partie seulement du périmètre autorisé
La réglementation européenne, récemment révisée, renforce les « exigences générales en matière de sécurité et de performances » 496 ( * ) relatives aux dispositifs médicaux. Le marquage CE vise à attester du respect desdites exigences pour un certain nombre de « destinations », présentées par le fabricant dans l'évaluation clinique préalable 497 ( * ) .
En cohérence, l'inscription sur la LPPR peut être subordonnée « au respect de spécifications techniques, d'indications thérapeutiques ou diagnostiques et de conditions particulières de prescription, d'utilisation et de distribution » 498 ( * ) .
Or d'après l'étude d'impact jointe au PLFSS, « il arrive que certains industriels sollicitent une inscription d'un dispositif à la LPP sur un périmètre plus restreint que ce qu'aurait permis leurs études cliniques réalisées dans le champ de leur marquage CE [...] , avec le souhait de maximiser le prix qu'ils peuvent obtenir en retenant une population de niche » 499 ( * ) .
Le prix des dispositifs médicaux, fixé conventionnellement avec le CEPS ou par décision de ce dernier, tient compte, en effet, de l'amélioration du service rendu ou attendu comme des volumes de vente anticipés 500 ( * ) .
b) L'article 31 vise à inciter les exploitants à demander l'inscription sur l'ensemble du périmètre autorisé par le marquage CE
Sur le modèle du dispositif prévu par l'article 30 du PLFSS pour le médicament, il est proposé d'inciter les exploitants à couvrir entièrement le périmètre du marquage CE en appliquant, dans le cas contraire, une remise obligatoire à leur produit.
À cette fin, le 15° du I insère dans le code de la sécurité sociale un nouvel article L. 165-4-2, qui prévoit que lorsqu'un dispositif médical est, à la demande expresse de l'exploitant, inscrit sur la LPPR pour un périmètre d'indications plus restreint que celui dans lequel ce dispositif présente un service attendu suffisant, l'exploitant est contraint de verser des remises. Celles-ci sont dues jusqu'à l'inscription du dispositif pour l'ensemble des indications concernées.
Le CEPS déterminera le montant des remises sur la base d'un barème fixé par arrêté en fonction des tailles respectives des populations cibles des indications concernées et de celles pour lesquelles le dispositif est inscrit.
2. Améliorer les contrôles sur les dispositifs inscrits sur ligne générique
a) L'inscription sur ligne générique de la LPPR
Aux termes de l'article L. 165-1 du code de la sécurité sociale, deux modalités d'inscription sur la LPPR coexistent .
Un produit peut, d'une part, être inscrit « sous forme de marque ou de nom commercial » : dans ce cas, correspondant aux produits princeps , l'exploitant dépose un dossier de demande de remboursement, et la CNEDiMTS évalue les niveaux de service attendu et d'amélioration du service attendu fondant la décision d'inscription comme la valorisation du tarif de responsabilité.
Les dispositifs peuvent également, d'autre part, être inscrits « par la description générique de tout ou partie du produit concerné » : dans ce cas, c'est l'exploitant lui-même qui appose sur son dispositif marqué CE le code LPPR correspondant à la ligne générique à laquelle il estime que son produit appartient, le dispositif ne faisant l'objet d'aucune évaluation par la CNEDiMTS. L'inscription sous description générique donne accès au tarif de remboursement associé à la ligne concernée.
Les exploitants et distributeurs au détail sont toutefois tenus de déclarer auprès de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) l'ensemble des produits ou prestations qu'ils commercialisent et inscrivent sur la LPPR, y compris par description générique, en précisant pour chaque produit ou prestation le code correspondant à l'inscription sur la liste 501 ( * ) .
b) L'article 31 vise à renforcer le contrôle des dispositifs des lignes génériques
Pour améliorer les contrôles visant les produits et prestations inscrits sur ligne générique, simplifier les démarches des entreprises et « renforcer la lutte contre la fraude » 502 ( * ) , l'article 31 substitue à l'actuelle déclaration auprès de l'ANSM un contrôle par la Cnam du respect des spécifications techniques attachées aux lignes de la LPPR.
Pour ce faire, le 16° du I abroge l'article L. 165-5 du code de la sécurité sociale encadrant la procédure de déclaration à l'ANSM. Le 17° du I insère au sein du code de la sécurité sociale un nouvel article L. 165-5-1-1 prévoyant que le directeur général de la Cnam peut à tout moment procéder, ou faire procéder sous son autorité par des organismes compétents désignés par arrêté, au contrôle du respect des spécifications techniques auxquelles l'inscription sur la LPPR est subordonnée.
Lorsque ce contrôle révèle un manquement, le directeur général :
- en informe, après avoir mis l'exploitant en mesure de présenter ses observations, les ministres chargés de la santé et de la sécurité sociale sans délai, les ministres l'informant en retour de toute mesure prise ;
- met l'exploitant en demeure de rembourser la somme correspondant, le cas échéant, au montant remboursé à tort par l'assurance maladie ;
- peut prononcer, en fonction de la gravité des faits reprochés, une pénalité financière à l'encontre de l'exploitant, dans la limite de 10 % de son chiffre d'affaires hors taxe réalisé en France au titre du dernier exercice clos et pour le produit considéré.
3. Renforcer l'évaluation post-inscription
a) L'inscription sur la LPPR et la prise en charge des dispositifs peut être assortie d'une obligation d'évaluation post-inscription
Les exploitants ou distributeurs au détail de dispositifs inscrits à la LPPR peuvent devoir recueillir, postérieurement à l'inscription, des données relatives aux performances, à la sécurité, ou au comportement médico-économique du produit en vie réelle.
L'avis rendu par la CNEDiMTS en vue de l'inscription d'un dispositif sur la LPPR peut requérir la réalisation d'études complémentaires nécessaires à l'évaluation du service rendu ou de son amélioration 503 ( * ) . Les résultats de ces études devront être fournis à la commission lors du plus prochain renouvellement d'inscription 504 ( * ) .
Par ailleurs, l'accord-cadre conclu entre le CEPS et les syndicats ou organisations regroupant les exploitants et distributeurs peut également définir les conditions et modalités de mise en oeuvre de telles études post-inscription 505 ( * ) . Celles-ci peuvent être déclinées dans les conventions conclues entre le comité et l'exploitant ou le distributeur.
Enfin, la prise en charge des dispositifs médicaux inscrits sur la LPPR ou remboursés aux hôpitaux au titre des groupes homogènes de séjour (GHS) peut être subordonnée au recueil et à la transmission d'informations relatives aux patients traités, au contexte de la prescription, aux indications dans lesquelles le produit ou la prestation est prescrit et aux résultats ou effets de ces traitements 506 ( * ) .
D'après l'étude d'impact, les exploitants, distributeurs et professionnels de santé ne recueillent toutefois pas toujours les données de vie réelles requises en application de ces dispositions.
b) L'article 31 vise à améliorer le recueil des données nécessaires aux évaluations post-inscription
Le 3° du I modifie l'article L. 162-17-1-2 du code de la sécurité sociale pour étendre l'obligation de recueil de données en vie réelle des établissements de santé aux actes et dispositifs « nécessitant un encadrement spécifique pour des raisons de santé publique ou susceptibles d'entraîner des dépenses injustifiées », au sens de l'article L. 1151-1 du code de la santé publique.
Le 14° du I complète l'article L. 165-4-1 du code de la sécurité sociale encadrant les conventions conclues par le CEPS en prévoyant que, lorsque les conventions signées avec un exploitant ou un distributeur au détail prévoient la réalisation d'études postérieures à l'inscription du produit ou de la prestation sur la LPPR, l'absence de transmission des résultats dans les délais impartis par la convention fait obstacle à la poursuite de la prise en charge du produit ou de la prestation, sauf versement de remises par l'exploitant ou le distributeur de détail concerné.
Enfin, le b) du 9° du I modifie L. 165-1-5 du même code relatif à la prise en charge transitoire des dispositifs n'étant pas encore inscrits pour prévoir que celle-ci est subordonnée au respect, par l'exploitant, d'un protocole de recueil, à sa charge, des données défini par la CNEDiMTS.
C. Adapter le régime de prise en charge transitoire et l'encadrement de la télésurveillance
Enfin, l'article 31 apporte aux dispositifs de prise en charge transitoire et de financement de la télésurveillance des adaptations mineures.
1. Compléter le dispositif de prise en charge transitoire
a) Le régime de prise en charge transitoire permet l'accès aux dispositifs innovants n'étant pas encore inscrits sur la LPPR
Créé par la LFSS pour 2019507 ( * ) sur le modèle de la procédure des autorisations temporaires d'utilisation (ATU) existant pour le médicament, le dispositif de prise en charge transitoire (PECT) permet le remboursement par l'assurance maladie de dispositifs médicaux qui ne sont pas encore inscrits sur la LPPR508 ( * ). Seuls certains établissements de santé peuvent distribuer ce produit ou cette prestation et bénéficier de sa prise en charge.
La PECT vise les dispositifs innovants mais disposant, toutefois, d'évaluations cliniques ayant permis leur marquage CE . La demande est adressée par l'exploitant aux ministres chargés de la santé et de la sécurité sociale, qui peuvent autoriser la prise en charge après avis de la CNEDiMTS. L'industriel s'engage à déposer, dans un délai de douze mois à compter de sa demande, une demande d'inscription sur la LPPR.
Le montant de la prise en charge est négocié : l'exploitant fait une proposition de compensation maximale aux ministres compétents, qui peuvent s'y opposer et, dans ce cas, formuler une contre-offre. Lorsqu'un produit ayant fait l'objet d'une PECT est inscrit sur la LPPR, une convention conclue avec le CEPS détermine le prix net de référence du produit ou de la prestation. Si celui-ci s'avère inférieur à la compensation maximale perçue pendant la période de PECT, l'exploitant reverse la différence entre le chiffre d'affaires facturé aux établissements de santé et celui qui aurait résulté du prix net de référence fixé, sous forme de remises.
La PECT ne couvre pas les actes associés à l'utilisation du produit , qui demeurent pris en charge sur inscription dans la classification commune des actes médicaux (CCAM)509 ( * ).
b) L'article 31 vise à étendre la prise en charge transitoire des dispositifs médicaux aux actes associés
Le c) du 9° du I modifie l'article L. 165-1-5 du code de la sécurité sociale, relatif à la PECT, pour prévoir :
- que lorsque l'utilisation d'un produit bénéficiant de la PECT est liée à un acte qui n'est pas prévu par la CCAM, un arrêté pris après avis de la HAS peut procéder à l'inscription transitoire de cet acte dans la classification commune pour la durée de la PECT ;
- que cet arrêté prévoit le montant de la compensation financière versée pour la réalisation de cet acte ;
- que lorsque le produit ayant fait l'objet de la PECT est inscrit sur la LPPR, l'inscription transitoire de l'acte est prolongée jusqu'à son inscription dans la CCAM.
En conséquence, le a) du 9° du I précise au même article que l'avis de la CNEDiMTS préalable à l'autorisation de la prise en charge transitoire d'un produit décrit, le cas échéant, les actes associés à son utilisation.
Enfin, le 18° du I précise à l'article L. 165-1-5 du code de la sécurité sociale que les produits bénéficiant de la PECT sont pris en charge en sus des prestations d'hospitalisations, soit hors GHS.
2. Compléter et reporter le régime de prise en charge de la télésurveillance
a) Des modalités de prise en charge pérennes de la télésurveillance ont été définies par la LFSS pour 2022
La télésurveillance fait partie, aux côtés de la téléconsultation ou de la téléexpertise, des actes de télémédecine. Elle a pour objet de permettre à un professionnel médical de surveiller et d'interpréter à distance les données nécessaires au suivi médical d'un patient , transmises au moyen d'un dispositif médical numérique (DMN) 510 ( * ) .
La LFSS pour 2014 511 ( * ) a permis le lancement des premières expérimentations de déploiement de la télémédecine, prolongées jusqu'en 2021 512 ( * ) dans le cadre du programme « expérimentations de télémédecine pour l'amélioration des parcours de santé » (ETAPES) , centré sur la prise en charge de quatre pathologies chroniques : le diabète et les insuffisances cardiaque, rénale et respiratoire.
Enfin et conformément à un engagement du Ségur de la santé de l'été 2020 513 ( * ) , la LFSS pour 2022 a inscrit dans le droit commun un régime pérenne de prise en charge de la télésurveillance 514 ( * ) .
Désormais, le code de la sécurité sociale prévoit les conditions de prise en charge des activités de télésurveillance médicale :
- celles-ci doivent être inscrites sur une liste spécifique, après avis de la CNEDiMTS 515 ( * ) ;
- la surveillance médicale doit être assurée par un opérateur de télésurveillance médicale, ayant déclaré ses activités à l'agence régionale de santé 516 ( * ) ;
- l'opérateur de télésurveillance médicale, professionnel médical, doit avoir mis à la disposition de l'assuré le dispositif médical numérique au moyen duquel la surveillance médicale est exercée 517 ( * ) ;
- enfin, la prise en charge est subordonnée à l'utilisation effective du DMN par le patient, les données permettant de satisfaire à ce contrôle pouvant être transmises par les opérateurs de télésurveillance, avec l'accord du patient 518 ( * ) .
La télésurveillance, mêlant dispositif médical et acte de soins, est rémunérée par un montant forfaitaire fixé par arrêté , qui tient compte des moyens humains nécessaires à la surveillance médicale et des caractéristiques des dispositifs médicaux numériques impliqués 519 ( * ) .
La LFSS pour 2022 disposait enfin que ces dispositions entreraient en vigueur à une date fixée par décret, et au plus tard le 1 er juillet 2022 et que les dispositifs médicaux de télésurveillance médicale seraient, en conséquence, radiés de la LPPR au plus tard le 1 er janvier 2023.
b) L'article 31 vise à apporter quelques adaptations à ce régime de prise en charge
Le 5° du I modifie l'article L. 162-56 du code de la sécurité sociale pour prévoir que les données d'utilisation du DMN peuvent être transmises non pas seulement par les opérateurs de télésurveillance, mais également par l'exploitant du DMN.
Le II de l'article 31 modifie l'article 1 635 bis AH du code général des impôts pour prévoir que l'inscription d'une activité de télésurveillance sur la liste spécifique est, comme l'est l'inscription sur la LPPR, subordonnée au paiement d'un droit au profit de la Cnam fixé par arrêté dans la limite de 5 600 euros.
Enfin, les IV et V reportent l'entrée en vigueur de la réforme en modifiant :
- la LFSS pour 2022, pour reporter au 1 er janvier 2023 la radiation des dispositifs médicaux de télésurveillance médicale de la LPPR, prévue initialement le 1 er janvier 2023 au plus tard ;
- la LFSS pour 2018, pour prolonger l'expérimentation ETAPES jusqu'au 30 juin 2023 au plus tard et prévoir que les expérimentateurs continuent de bénéficier de cette prise en charge financière sous réserve d'un engagement à déposer une demande d'inscription à la nouvelle liste spécifique pour le 31 janvier 2023.
L'étude d'impact fait état d'un report nécessaire « pour assurer une transition fluide sur le plan réglementaire et technique » 520 ( * ) .
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Dans le texte sur lequel le Gouvernement a engagé sa responsabilité en application de l'article 49, alinéa 3, de la Constitution, l'article 31 n'a subi que des modifications rédactionnelles.
III - La position de la commission
La commission a une nouvelle fois regretté que l'article rassemble de trop nombreuses mesures disparates, à la portée très inégale. Cette agrégation ne favorise ni la lisibilité du texte, ni la clarté des débats.
A. Sur la réforme de la tarification
La réforme des modalités de tarification des produits et prestations de la LPPR doit sans doute être tenue pour le coeur de ces dispositions. À cet égard, la commission a favorablement accueilli les mesures visant à mieux distinguer la tarification des produits de la rémunération des distributeurs et des prestations associées. Elle a formulé le voeu que la réforme permette à de rendre plus transparent le partage de la valeur dans le secteur. Elle souhaite également que cette mesure conduise à valoriser plus justement l'activité de chacun des acteurs et, notamment, l'apport des prestations.
En revanche, la commission s'est interrogée sur les modalités de mise en oeuvre de la réforme des marges de distribution .
La loi prévoit la fixation, par voie réglementaire, des marges de distribution des dispositifs médicaux inscrits sur la LPPR, « dans des conditions et à des dates fixées par décret, et au plus tard le 31 décembre 2025, en fonction des catégories de produits ou prestations concernées ». Le calendrier n'est pas davantage précisé.
Les objectifs de la réforme apparaissent également incertains. Celle-ci est tour à tour présentée, dans l'étude d'impact, comme un moyen « garantir une marge cohérente pour les distributeurs au détail » puis comme une source d'économies importantes, devant permettre de réduire les dépenses de l'assurance maladie de cinquante à cent millions d'euros par an. Interrogé à ce sujet par la rapporteure, le ministère a souligné l'existence de marges de distribution aujourd'hui très élevées dans certains secteurs, sans fournir à l'appui de cette affirmation de données suffisamment précises pour emporter l'adhésion.
Enfin, la commission souligne que la très grande hétérogénéité des acteurs participant à la distribution des dispositifs médicaux inscrits sur la LPPR rend malaisée l'estimation du périmètre et des conséquences de cette réforme. L'étude d'impact ne cite, parmi les distributeurs « de ville », que les prestataires de services et distributeurs de matériel (PSDM) et les pharmacies d'officine. Les opticiens ou les audioprothésistes, par exemple, ne sont pas mentionnés. La mesure a pourtant suscité, au sein de ces professions par ailleurs concernées par la réforme du 100 % santé, d'importantes inquiétudes.
B. Sur les autres mesures portées par l'article
Si la commission s'est montrée favorable aux mesures visant à mieux réguler le périmètre des demandes d'inscription sur la LPPR et à inciter à la réalisation des évaluations post-inscription, elle a toutefois souligné la nécessité de veiller à ce que les coûts induits pour les exploitants demeurent proportionnés aux objectifs poursuivis.
À cet égard, le transfert des contrôles réalisés sur les inscriptions sur lignes génériques de la LPPR à la Cnam, permettant de supprimer une obligation déclarative désormais inutile pour les entreprises, lui a paru souhaitable.
Elle soutient, enfin, l'extension du dispositif de prise en charge transitoire devant permettre d'y inclure les actes associés. Celle-ci devrait permettre d'améliorer l'effectivité du dispositif et, partant, l'accès aux dispositifs innovants des assurés.
La commission vous demande d'adopter cet article sans modification.
Article 31 bis
(nouveau)
Possibilité de substitution de certains dispositifs
médicaux par le pharmacien d'officine
Cet article, inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité en application du troisième alinéa de l'article 49 de la Constitution, vise à autoriser et encadrer la substitution de certains dispositifs médicaux par le pharmacien d'officine.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
A. L'état du droit
En principe, la dispensation est subordonnée à la prescription : l'article L. 5125-23 du code de la santé publique dispose que, en l'absence d'urgence, « Le pharmacien ne peut délivrer un médicament ou produit autre que celui qui a été prescrit, ou ayant une dénomination commune différente de la dénomination commune prescrite, qu'avec l'accord exprès et préalable du prescripteur ».
• Toutefois, pour le médicament et dans l'objectif notamment de maîtriser les dépenses d'assurance maladie, la loi a progressivement ouvert, depuis 1999, la possibilité au pharmacien, à la condition que le prescripteur ne l'ait pas exclu par une mention expresse, de délivrer par substitution à la spécialité prescrite 521 ( * ) :
- une spécialité du même groupe générique , soit disposant de la même composition qualitative et quantitative en principes actifs, de la même forme pharmaceutique et dont la bioéquivalence est démontrée par des études appropriées 522 ( * ) ;
- ou, dans les cas listés par arrêté, une spécialité du même groupe hybride , soit une spécialité ne répondant pas à la définition d'une spécialité générique, mais qualifiée ainsi après des essais précliniques et cliniques appropriés et déterminés en fonction des différences observées avec la spécialité de référence 523 ( * ) .
Lorsqu'un pharmacien délivre un médicament par substitution, il doit inscrire le nom de la spécialité qu'il a délivrée.
• Une faculté de substitution d'un dispositif médical similaire au dispositif prescrit a exceptionnellement été ouverte aux pharmaciens et distributeurs dans le contexte de la crise sanitaire.
Deux arrêtés des 23 mars 524 ( * ) et 10 juillet 2020 525 ( * ) ont ainsi successivement permis, en cas de rupture avérée d'un dispositif médical nécessaire à la continuité des soins d'un patient dont l'interruption pourrait être préjudiciable à sa santé, de substituer au dispositif médical indisponible un autre dispositif médical répondant à quatre critères :
- avoir un usage identique à celui du dispositif médical substitué ;
- disposer de spécifications techniques équivalentes à celles du dispositif médical substitué ;
- être inscrit sur la liste des produits et prestations remboursables (LPPR) ;
- ne pas entraîner de dépenses supplémentaires pour le patient et l'assurance maladie.
Ces dispositions ont depuis été abrogées 526 ( * ) .
B. Le dispositif proposé
Le présent article, issu d'un amendement de M. Bertrand Bouyx conservé dans le texte sur lequel le Gouvernement a engagé sa responsabilité, vise à inscrire dans le code de la santé publique un régime pérenne de délivrance de dispositifs médicaux par substitution.
À cette fin, le I de l'article 31 bis insère dans le code un nouvel article L. 5125-23-3 prévoyant que, par dérogation au principe posé à l'article L. 5125-23, le pharmacien peut délivrer, par substitution au produit prescrit, un produit comparable lorsque quatre conditions sont remplies :
- le produit figure sur une liste fixée par arrêté pris après avis de la Haute Autorité de santé (HAS) ;
- le cas échéant, les conditions de substitution et d'information du prescripteur et du patient à l'occasion de cette substitution fixées par ladite liste peuvent être respectées ;
- le prescripteur n'a pas exclu la possibilité de cette substitution par une mention expresse et justifiée portée sur l'ordonnance, tenant à la situation médicale du patient ;
- si le produit prescrit figure sur la LPPR, cette substitution s'effectue dans les conditions prévues à l'article L. 162-16 du code de la sécurité sociale.
Le cas échéant, le pharmacien inscrit le nom du produit délivré sur l'ordonnance et informe le prescripteur et le patient de la substitution.
En outre, le II insère à l'article L. 162-16 du code de la sécurité sociale un V bis prévoyant :
- que les dispositifs médicaux dispensés par un pharmacien d'officine en application de l'article L. 5125-23-3 sont pris en charge par l'assurance maladie lorsqu'ils sont inscrits sur la LPPR ;
- que, le cas échéant, la substitution ne doit pas entraîner une dépense supplémentaire pour l'assurance maladie supérieure à la dépense qu'aurait entraînée la délivrance du produit prescrit.
II - La position de la commission
Dans la mesure où elle est susceptible d'améliorer l'accès de certains patients aux dispositifs médicaux et de contribuer à la maîtrise des dépenses relatives aux produits de santé, la commission est favorable à la faculté de substitution prévue au présent article.
La commission vous demande d'adopter cet article sans modification.
Article 31 ter
(nouveau)
Élargissement du 100 % santé aux
prothèses capillaires
Cet article, inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité en application du troisième alinéa de l'article 49 de la Constitution, propose des adaptations légales pour permettre à l'assurance maladie d'étendre le 100 % santé aux prothèses capillaires.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
A. L'état du droit
• Prévu par l'article 51 de la LFSS pour 2019, le dispositif du 100 % santé permet, depuis le 1 er janvier 2020, une suppression intégrale des restes à charge des patients sur un panier défini pour les soins dentaires prothétiques, les dispositifs d'optique médicale et les aides auditives 527 ( * ) . Le financement de cette prise en charge est partagé entre les assurances maladies obligatoires (AMO) et les complémentaires (AMC).
La commission a souhaité, en application de l'article L.O. 132- 3-1 du code des juridictions financière, recourir à l'expertise de la Cour des comptes afin que soit dressé un premier bilan de la réforme du 100 % santé 528 ( * ) . La Cour a ainsi montré que l'objectif d'amélioration de l'accès aux soins visé avait été atteint grâce à un plafonnement des prix très largement respecté . Toutefois, elle a relevé que la pénétration du panier de soins 100 % remboursé s'avérait très inégale selon les secteurs avec des résultats positifs sur les prothèses dentaires et les aides auditives mais décevants pour l'optique.
• Les prothèses capillaires, permettant de masquer l'alopécie provoquée par une maladie ou un traitement médical, sont prises en charge par l'Assurance maladie selon des tarifs de responsabilité et des prix limite de vente au public fixés, en l'absence d'accord conventionnel, par décision du comité économique des produits de santé 529 ( * ) . Le prothésiste capillaire doit être agréé par l'assurance maladie.
Cette prise en charge varie selon que la prothèse capillaire appartient à une classe 1, intégralement prise en charge par l'AMO, ou une classe 2, pour laquelle le reste à charge moyen constaté en 2020 est de 376 euros pour une prise en charge par l'Assurance maladie de 250 euros. Les prothèses de classe 1 ne sont faites que de cheveux synthétiques tandis que celles de classe 2, que 60 % des patients choisissent, doivent comporter au moins 30 % de cheveux naturels.
Selon la Cnam, dans son rapport « Charges et produits » de juillet 2022, les dépenses de prise en charge des prothèses capillaires ne représentaient que 15 millions d'euros de remboursements pour l'Assurance maladie en 2021.
B. Le dispositif proposé : inclure les prothèses capillaires dans le panier de soins du 100 % santé
La LFSS pour 2019 avait inséré l'article L. 165-1-4 du code de la sécurité sociale afin de prévoir la possibilité d'imposer aux distributeurs ou fabricants :
- de proposer et de disposer des produits appartenant aux classes à prise en charge renforcée ou offres « 100 % santé » ;
- de participer à un dispositif d'évaluation portant à la fois sur la qualité de la prise en charge et la satisfaction du patient, et la mise en oeuvre conforme des offres « 100 % santé ».
Si le distributeur refuse ces dispositions, l'ensemble des produits et prestations qu'il distribue ne peuvent alors être admis au remboursement.
• Le présent article modifie l'article L. 165-1-4 afin de prévoir un régime adapté à une extension du dispositif 100 % santé aux prothèses capillaires .
Si les prothèses capillaires peuvent être vendues par les pharmaciens d'officine, d'autres exploitants ou distributeurs au détail n'appartiennent pas aux professions réglementées par le code de la santé publique (coiffeurs, perruquiers).
Dans ce cas, pour qu'un vendeur propose des prothèses capillaires incluses dans le 100 % santé, le dispositif proposé insère une obligation légale faites à l'exploitant ou le distributeur au détail de posséder un identifiant de facturation délivré par les organismes d'assurance maladie lui permettant d'établir des feuilles de soins susceptibles d'être présentées au remboursement des produits ou prestations.
Il convient de noter que le régime serait ainsi adapté mais que les termes de prothèses capillaires ne seraient pas littéralement inscrits à l'article L. 871-1 du code de la sécurité sociale. Ils seraient inclus dans les « dispositifs médicaux à usage individuel admis au remboursement » des AMC y compris en sus des tarifs de responsabilité (offre 100 % santé).
II - La position de la commission : une mesure bénéfique qui devra s'accompagner d'évaluation générale des paniers de soins retenus pour le 100 % santé
À la lumière des remarques formulées par la Cour des comptes, la commission notait en juillet 2022 qu'une « extension du dispositif [du 100 % santé] apparai [ssait] risquée à court terme, du fait des difficultés de suivi, de pilotage et d'évaluation relevées par la Cour dans son rapport » 530 ( * ) . La Cour fixait en effet comme préalable à des éventuels enrichissements une évaluation en profondeur des paniers actuels. La rapporteure ne peut que regretter par ailleurs que le présent article ait été inséré par amendement, déposé notamment par le Gouvernement, et ne soit pas ainsi étayé par une étude d'impact.
Le rapport « charges et produits » précité évoque cependant l'élargissement du panier de soins du dispositif 100 % santé. Selon la Cnam, de toutes les extensions envisageables, les prothèses capillaires apparaissent comme les dispositifs les plus facilement intégrables à court terme. Il est toutefois difficile de prévoir une incidence financière précise compte tenu des paramètres détaillés qu'il conviendra de déterminer.
La caisse note qu'« une enveloppe de 20 millions d'euros pourrait constituer une première évaluation, à deux-tiers supportée par l'AMO, pour un tiers par les assureurs complémentaires ».
En tout état de cause, l'intégration des prothèses capillaires au dispositif du 100 % santé ne peut qu'améliorer l'accès à ces prothèses en raison de l'important renoncement souligné par la Cnam. En 2020, 347 000 personnes étaient traitées par chimiothérapie dont 4 0% pour un cancer du sein ou hématologique. Or, seuls 50 114 patients ont bénéficié d'une prise en charge au titre d'une prothèse capillaire en 2021.
La rapporteure constate que l'inclusion proprement dite des prothèses capillaires dans le dispositif 100 % santé ne nécessiterait que des textes règlementaires ainsi que, le cas échéant, des adaptations du remboursement, du prix limite de vente au public ou enfin des améliorations de la qualité technique des prothèses. Le présent article se borne à adapter le régime légal afin de pouvoir imposer aux prothésistes non réglementées, coiffeurs ou perruquiers, la vente de prothèses éligibles à l'offre du 100 % santé. La rapporteure émet par ailleurs quelques doutes sur la recevabilité de l'article au regard du domaine des lois de financement de la sécurité sociale.
Sous les réserves formulées d'amélioration, recommandée par la Cour des comptes, de l'évaluation et du suivi du dispositif 100 % santé, la commission est favorable à l'extension du panier de soins aux prothèses capillaires et souscrit au présent article.
La commission vous demande d'adopter cet article sans modification.
* 432 Article 78 du PLFSS pour 2021.
* 433 Article 58 du PLFSS pour 2022.
* 434 Article L. 5121-12 du code de la santé publique.
* 435 Article D. 5121-69-3 du code de la santé publique.
* 436 Article R. 5121-68 du code de la santé publique.
* 437 Article R. 5121-69 du code de la santé publique.
* 438 Article R. 5121-69-2 du code de la santé publique.
* 439 Article L. 162-16-5-1 du code de la sécurité sociale.
* 440 Article L. 162-16-5-1-1 du code de la sécurité sociale.
* 441 HAS, ANSM, Autorisation d'accès précoce aux médicaments : un premier bilan positif et des principes d'évaluation affinés , 20 mai 2022
* 442 Article L. 5121-12-1 du code de la santé publique.
* 443 Article L. 162-16-5-2 du code de la sécurité sociale.
* 444 Annexe n° 9 du PLFSS pour 2023, p. 197.
* 445 Annexe n° 9 du PLFSS pour 2023, p. 197.
* 446 Règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) n° 726/2004.
* 447 Article 2 du règlement (CE) n° 1397/2007.
* 448 Partie IV de l'annexe I de la directive 2001/83/CE du Parlement et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain.
* 449 Partie IV de l'annexe I de la directive 2001/83/CE précitée.
* 450 Article L. 162-22-7 du code de la sécurité sociale.
* 451 Annexe n° 9 du PLFSS pour 2023, p. 196.
* 452 Commission des comptes de la sécurité sociale, Les comptes de la sécurité sociale : résultats 2021 et prévisions 2022-2023 , tome 1, pp. 71-72.
* 453 CEPS, Rapport d'activité 2020 , décembre 2021, p. 25.
* 454 Article L. 162-16-4 du code de la sécurité sociale.
* 455 Article L. 162-16-5 du code de la sécurité sociale.
* 456 Article L. 162-16-6 du code de la sécurité sociale.
* 457 Article L. 162-16-4 du code de la sécurité sociale.
* 458 Article 98 de la LFSS pour 2017.
* 459 CEPS, Rapport d'activité 2019 , septembre 2020.
* 460 CEPS, Rapport d'activité 2020 , décembre 2021.
* 461 Ibid .
* 462 Article L. 162-18 du code de la sécurité sociale.
* 463 Articles L. 162-16-5-1-1 et L. 162-16-5-2 du code de la sécurité sociale.
* 464 L. 138-13 du code de la sécurité sociale : voir commentaire de l'article 9 bis.
* 465 Article L. 162-18 du code de la sécurité sociale.
* 466 Annexe n° 9 du PLFSS pour 2023, p. 199.
* 467 Ibid ., p. 195.
* 468 Ibid .
* 469 Loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 470 Article L. 5111-4 du code de la santé publique.
* 471 Article L. 5121-29 du code de la santé publique.
* 472 Article R. 5124-49-4 du code de la santé publique modifié par le décret n° 2021-349 du 30 mars 2021.
* 473 Article L. 5121-31 du code de la santé publique.
* 474 Article L. 5121-32 du code de la santé publique.
* 475 Par renvoi à l'article L. 162-37 du code de la sécurité sociale, affectant à la Cnam certaines remises versées par les entreprises.
* 476 Article L. 5126-1 du code de la santé publique.
* 477 Article 78 de la loi n° 2020-1525 du 7 décembre 2020 d'accélération et de simplification de l'action publique.
* 478 Avant l'intervention de la loi n° 2020-1525, le ministre chargé de la santé arrêtait cette liste directement.
* 479 Article L. 5126-6 du code de la santé publique.
* 480 Premier alinéa de l'article L. 162-17 du code de la sécurité sociale.
* 481 Article 43 de la n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020.
* 482 Voir le magazine de l'Inserm, « Cannabis médical : un écran de fumée ? », sur https://www.inserm.fr/actualite/cannabis-medical-un-ecran-de-fumee/
* 483 Article 2 du règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE.
* 484 Ibid .
* 485 Article L. 165-1 du code de la sécurité sociale.
* 486 Article L. 165-2 du code de la sécurité sociale.
* 487 Annexe n° 9 au PLFSS, p. 214.
* 488 Article L. 162-38 du code de la sécurité sociale.
* 489 Arrêté du 4 août 1987 relatif aux prix et aux marges des médicaments remboursables et des vaccins et des allergènes préparés spécialement pour un individu modifié.
* 490 Article L. 162-38 du code de la sécurité sociale.
* 491 Lettre d'orientation ministérielle du 17 août 2016, citée par le CEPS dans son rapport annuel pour 2020 (p. 82).
* 492 Même article L. 165-1-1-1 du code de la sécurité sociale.
* 493 Article L. 165-2-2 du code de la sécurité sociale.
* 494 Articles L. 165-2 et L. 165-3 du code de la sécurité sociale.
* 495 Article L. 165-4 du code de la sécurité sociale.
* 496 Désormais listées à l'annexe I du règlement (UE) 2017/745 du Parlement européen et du Conseil précité.
* 497 La destination est définie à l'article 2 de la directive comme « l'utilisation à laquelle un dispositif est destiné d'après les indications fournies par le fabricant sur l'étiquette, dans la notice d'utilisation ou dans les documents ou indications publicitaires ou de vente, et comme celles présentées par le fabriquant dans l'évaluation clinique ».
* 498 Article L. 165-1 du code de la sécurité sociale.
* 499 Annexe n° 9 au PLFSS, p. 215.
* 500 Article L. 165-2 du code de la sécurité sociale.
* 501 Article L. 165-5 du code de la sécurité sociale.
* 502 Annexe n° 9 au PLFSS, p. 215.
* 503 Article R. 165-11 du code de la sécurité sociale.
* 504 Article R. 165-11-1 du code de la sécurité sociale.
* 505 Article L. 165-4-1 du code de la sécurité sociale.
* 506 Article L. 162-17-1-2 du code de la sécurité sociale.
* 507 Article 65 de la LFSS pour 2019.
* 508 Article L. 165-1-5 du code de la sécurité sociale.
* 509 Article L. 162-1-7 du code de la sécurité sociale.
* 510 Article L. 162-48 du code de la sécurité sociale.
* 511 Article 36 de la LFSS pour 2014.
* 512 L'expérimentation, initialement d'une durée de quatre ans, a été prolongée par l'article 91 de la LFSS pour 2017 puis par l'article 54 de la LFSS pour 2018.
* 513 La mesure 24 comprenait un engagement à « fixer le périmètre et les principes du financement de la télésurveillance puis confier aux partenaires conventionnels le soin de définir la rémunération afférente ».
* 514 Articles L. 162-1 à L. 162-1-23 du code de la sécurité sociale.
* 515 Article L. 162-52 du code de la sécurité sociale.
* 516 Article L. 162-51 du code de la sécurité sociale.
* 517 Article L. 162-49 du code de la sécurité sociale.
* 518 Article L. 162-56 du code de la sécurité sociale.
* 519 Article L. 162-54 du code de la sécurité sociale.
* 520 Annexe n° 9 au PLFSS, p. 217.
* 521 Article L. 5125-23 du code de la santé publique.
* 522 Article L. 5121-1 du code de la santé publique.
* 523 Ibid .
* 524 Arrêté du 23 mars 2020 prescrivant les mesures d'organisation et de fonctionnement du système de santé nécessaires pour faire face à l'épidémie de covid-19 dans le cadre de l'état d'urgence sanitaire.
* 525 Arrêté du 10 juillet 2020 prescrivant les mesures générales nécessaires pour faire face à l'épidémie de covid-19 dans les territoires sortis de l'état d'urgence sanitaire et dans ceux où il a été prorogé.
* 526 Arrêté du 1 er juin 2021 prescrivant les mesures générales nécessaires à la gestion de la sortie de crise sanitaire.
* 527 Auparavant, l'article L. 871-1 du code de la sécurité sociale se bornait à imposer aux contrats de complémentaire santé responsables et solidaires des garanties planchers et des plafonds de garanties applicables à certains postes de soin.
* 528 Cour des comptes, La réforme du 100 % santé : communication à la commission des affaires sociales du Séna t, juillet 2022.
* 529 Décision du 6 mars 2019 fixant le tarif de responsabilité et le prix limite de vente au public (PLV) en euros TTC des prothèses capillaires et des accessoires inscrits sur la liste prévue à l'article L. 165-1 du code de la sécurité sociale
* 530 Rapport d'information n° 832 (2021-2022) fait au nom de la commission des affaires sociales, sur l'enquête de la Cour des comptes sur la réforme du 100 % santé, par Mme Corinne Imbert.